
HULIO 40 mg-0,8 mL, solution injectable, boîte de 2 flacons ( nécessaires à injection) de 0,80 mL
Retiré du marché le : 18/07/2022
Dernière révision : 15/11/2021
Taux de TVA : 2.1%
Prix de vente : 467,80 €
Taux remboursement SS : 65%
Base remboursement SS : 467,80 €
Laboratoire exploitant : VIATRIS SANTE
Source :

Arthrite juvénile idiopathique
Arthrite juvénile idiopathique polyarticulaire
Hulio en association au méthotrexate est indiqué pour le traitement de l'arthrite juvénile idiopathique polyarticulaire évolutive chez les patients à partir de 2 ans en cas de réponse insuffisante à un ou à plusieurs traitements de fond (DMARD). Hulio peut être administré en monothérapie en cas d'intolérance au méthotrexate ou lorsque la poursuite du traitement par méthotrexate est inadaptée (pour l'efficacité en monothérapie, voir rubrique Propriétés pharmacodynamiques). L'adalimumab n'a pas été étudié chez les patients de moins de 2 ans.
Arthrite liée à l'enthésite
Hulio est indiqué pour le traitement de l'arthrite active liée à l'enthésite chez les patients à partir de 6 ans en cas de réponse insuffisante ou d'intolérance au traitement conventionnel (voir rubrique Propriétés pharmacodynamiques).
Psoriasis en plaques chez l'enfant et l'adolescent
Hulio est indiqué dans le traitement du psoriasis en plaques chronique sévère chez les enfants à partir de 4 ans et les adolescents en cas de réponse insuffisante à un traitement topique et aux photothérapies ou lorsque ces traitements sont inappropriés.
Hidrosadénite suppurée chez l'adolescent
Hulio est indiqué dans le traitement de l'hidrosadénite suppurée (maladie de Verneuil) active, modérée à sévère, chez les adolescents à partir de 12 ans en cas de réponse insuffisante au traitement systémique conventionnel de l'hidrosadénite suppurée (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Maladie de Crohn de l'enfant et l'adolescent
Hulio est indiqué dans le traitement de la maladie de Crohn active modérée à sévère, chez les enfants et les adolescents à partir de 6 ans, qui n'ont pas répondu à un traitement conventionnel comprenant un traitement nutritionnel de première intention et un corticoïde et/ou un immunomodulateur, ou chez lesquels ces traitements sont mal tolérés ou contre-indiqués.
Rectocolite hémorragique chez l'enfant et l'adolescent
Hulio est indiqué dans le traitement de la rectocolite hémorragique active, modérée à sévère chez les enfants et les adolescents à partir de 6 ans ayant eu une réponse inadéquate au traitement conventionnel, comprenant les corticoïdes et/ou la 6-mercaptopurine (6-MP) ou l'azathioprine (AZA), ou chez lesquels ces traitements sont mal tolérés ou contre-indiqués.
Uvéite chez l'enfant et l'adolescent
Hulio est indiqué dans le traitement de l'uvéite antérieure chronique non infectieuse chez les enfants et les adolescents à partir de 2 ans en cas de réponse insuffisante ou d'intolérance au traitement conventionnel ou pour lesquels un traitement conventionnel est inapproprié.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Tuberculose évolutive ou autres infections sévères telles que sepsis et infections opportunistes (voir rubrique Mises en garde spéciales et précautions d'emploi).
Insuffisance cardiaque modérée à sévère (NYHA classes III/IV) (voir rubrique Mises en garde spéciales et précautions d'emploi).
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, la dénomination du médicament et le numéro de lot du produit administré doivent être clairement enregistrés.
Infections
Les patients recevant des antagonistes du TNF sont plus prédisposés aux infections graves. Une fonction pulmonaire altérée peut augmenter le risque de développer des infections. Les patients doivent donc être surveillés étroitement afin de dépister des infections (y compris la tuberculose) avant, pendant et après le traitement par adalimumab. La durée d'élimination de l'adalimumab pouvant aller jusqu'à quatre mois, la surveillance devra être poursuivie pendant toute cette période.
Le traitement par Hulio ne doit pas être instauré tant que les infections évolutives, y compris les infections chroniques ou localisées, ne sont pas contrôlées. Chez les patients ayant été exposés à la tuberculose ou ayant voyagé dans des régions à haut risque de tuberculose ou de mycoses endémiques, par exemple histoplasmose, coccidioïdomycose ou blastomycose, les risques et bénéfices du traitement par Hulio doivent être pris en considération avant l'instauration du traitement (voir Autres infections opportunistes).
Les patients chez qui apparaît une nouvelle infection en cours de traitement par Hulio doivent faire l'objet d'une surveillance soigneuse et un bilan diagnostique complet doit être pratiqué. En cas d'apparition d'une nouvelle infection grave ou d'une septicémie, l'administration d'adalimumab doit être interrompue et un traitement antimicrobien ou antifongique approprié doit être instauré jusqu'à ce que l'infection soit contrôlée. Le médecin doit faire preuve de prudence avant d'utiliser Hulio chez des patients ayant des antécédents d'infection récidivante ou dans des conditions sous-jacentes susceptibles de les prédisposer aux infections, y compris un traitement concomitant par des médicaments immunosuppresseurs.
Infections graves
Des infections graves, incluant des septicémies dues à des infections bactériennes, mycobactériennes, fongiques invasives, parasitaires, virales ou à d'autres infections opportunistes, telles que listériose, légionellose et pneumocystose ont été rapportées chez des patients traités par l'adalimumab.
Les autres infections graves observées dans les essais cliniques sont : pneumonie, pyélonéphrite, arthrite septique et septicémie. Des cas d'infections nécessitant une hospitalisation ou ayant une issue fatale ont été rapportés.
Tuberculose
Des cas de tuberculose, incluant des cas de réactivation de la tuberculose et de primo-infection tuberculeuse, ont été rapportés pour des patients recevant de l'adalimumab. Des cas de tuberculoses pulmonaire et extra-pulmonaire (c'est-à-dire disséminée) ont été rapportés.
Avant l'instauration du traitement par Hulio, tous les patients doivent faire l'objet d'une recherche d'infection tuberculeuse active ou non (« latente »). Ce bilan doit comprendre une évaluation médicale détaillée chez les patients ayant des antécédents de tuberculose ou d'exposition antérieure possible à des patients atteints de tuberculose active et/ou d'un traitement immunosuppresseur actuel ou ancien. Des tests de dépistage appropriés (par exemple, test dermique à la tuberculine et radiographie pulmonaire) doivent être effectués chez tous les patients (conformément aux recommandations locales). Il est conseillé de noter la réalisation et les résultats de ces tests dans la carte de surveillance du patient. Il est rappelé aux prescripteurs que le test dermique à la tuberculine peut donner des faux- négatifs notamment chez les patients gravement malades ou immuno-déprimés.
En cas de diagnostic d'une tuberculose active, le traitement par Hulio ne doit pas être instauré (voir rubrique Contre-indications).
Dans toutes les situations décrites ci-dessous, il convient d'évaluer très attentivement le rapport bénéfice/risque du traitement.
En cas de suspicion d'une tuberculose latente, la consultation d'un médecin spécialiste, qualifié dans le traitement de la tuberculose, doit être envisagée.
En cas de diagnostic d'une tuberculose latente, une prophylaxie antituberculeuse appropriée et conforme aux recommandations locales doit être mise en œuvre avant le début du traitement par Hulio.
Une prophylaxie antituberculeuse doit également être envisagée avant l'instauration d'Hulio chez les patients ayant des facteurs de risque multiples ou significatifs de tuberculose malgré un test de dépistage de la tuberculose négatif et chez les patients ayant des antécédents de tuberculose latente ou active, chez qui l'administration d'un traitement antituberculeux approprié ne peut être confirmée.
Des cas de réactivation d'une tuberculose malgré un traitement prophylactique sont survenus chez des patients traités par l'adalimumab. Certains patients qui avaient été traités avec succès pour une tuberculose active ont développé à nouveau la maladie pendant le traitement par l'adalimumab.
Les patients devront être informés qu'il leur faudra consulter leur médecin en cas de survenue de signes ou symptômes évocateurs d'une infection tuberculeuse (par exemple, toux persistante, amaigrissement/perte de poids, fébricule, apathie), pendant ou après le traitement par Hulio.
Autres infections opportunistes
Des infections opportunistes, incluant des infections fongiques invasives, ont été observées chez des patients traités par l'adalimumab. Ces infections n'ont pas toujours été détectées chez les patients recevant des antagonistes du TNF, ce qui a retardé l'instauration d'un traitement approprié, avec parfois une issue fatale.
Chez les patients qui présentent des signes et symptômes tels que fièvre, malaise, perte de poids, sueurs, toux, dyspnée et/ou infiltrats pulmonaires ou une autre maladie systémique grave avec ou sans choc concomitant, une infection fongique invasive doit être suspectée ; dans ce cas, il convient d'arrêter immédiatement l'administration d'Hulio. Le diagnostic et la mise en place d'un traitement antifongique empirique chez ces patients doivent être effectués en accord avec un médecin ayant l'expérience de la prise en charge des patients ayant des infections fongiques invasives.
Réactivation d'hépatite B
Une réactivation d'hépatite B s'est produite chez des patients qui ont reçu un antagoniste du TNF y compris l'adalimumab et qui étaient porteurs chroniques de ce virus (c'est-à-dire, antigène de surface positif). Certains cas ont eu une issue fatale. Les patients doivent faire l'objet d'un dépistage d'infection à VHB avant l'instauration d'un traitement par adalimumab. Pour les patients pour lesquels le test de dépistage de l'hépatite B est positif, il est recommandé de consulter un médecin spécialisé dans le traitement de l'hépatite B.
Chez les porteurs du VHB qui nécessitent un traitement par adalimumab, il faut surveiller attentivement les signes et les symptômes d'infection active par le VHB tout au long du traitement et pendant plusieurs mois après son arrêt. Il n'y a pas de données disponibles suffisantes concernant le traitement de patients porteurs du VHB traités par antiviral pour prévenir une réactivation du VHB et traités par un antagoniste du TNF. Chez les patients qui développent une réactivation du VHB, Hulio doit être arrêté et un traitement antiviral efficace ainsi qu'un traitement complémentaire adapté doivent être instaurés.
Événements neurologiques
Les
antagonistes du TNF, dont l'adalimumab, ont été
associés dans de rares circonstances à l'apparition ou à l'exacerbation des
symptômes cliniques et/ou des signes radiologiques de maladies démyélinisantes du système nerveux central, y compris la
sclérose en plaques et la névrite optique, et de maladies démyélinisantes
périphériques, y compris le syndrome de Guillain-Barré. La prudence est
recommandée aux prescripteurs avant de traiter avec de l'adalimumab
les patients atteints d'une maladie démyélinisante du
système nerveux central ou périphérique, préexistante ou de survenue récente ;
l'arrêt du traitement doit être envisagé en cas d'apparition de l'un de ces
troubles.
L'association
entre l'uvéite intermédiaire et les affections démyélinisantes
du système nerveux central est connue. Une évaluation neurologique doit être
réalisée chez les patients présentant une uvéite intermédiaire non infectieuse
avant l'instauration du traitement par Hulio, et
répétée régulièrement au cours du traitement afin de rechercher toute maladie démyélinisante du système nerveux central préexistante ou
évolutive.
Réactions allergiques
Pendant les essais cliniques, les réactions allergiques graves associées à l'adalimumab ont été rares et des réactions allergiques non graves imputables à l'adalimumab ont été peu fréquentes. Des cas de réactions allergiques graves, incluant des réactions anaphylactiques, ont été rapportés après administration de l'adalimumab. En cas de survenue d'une réaction anaphylactique ou d'une autre réaction allergique grave, l'administration d'Hulio doit être immédiatement interrompue et un traitement approprié mis en œuvre.
Immunosuppression
Au cours d'une étude portant sur 64 patients atteints de polyarthrite rhumatoïde et traités par l'adalimumab, on n'a enregistré aucun élément évocateur d'une dépression de l'hypersensibilité de type retardé, d'une diminution des taux d'immunoglobulines ou d'une modification de la numération des lymphocytes effecteurs T et B, des lymphocytes NK, des monocytes/macrophages et des granulocytes neutrophiles.
Tumeurs malignes et troubles lymphoprolifératifs
Dans la partie contrôlée des essais cliniques avec des anti-TNF, il a été observé plus de cas de cancers y compris des lymphomes chez les patients traités par un anti-TNF que chez les patients du groupe contrôle. Cependant, l'incidence a été rare. Au cours de la surveillance post-marketing, des cas de leucémie ont été rapportés chez des patients traités par anti-TNF. De plus, il existe un contexte de risque accru de lymphome et de leucémie chez les patients atteints d'une polyarthrite rhumatoïde ancienne, inflammatoire et hautement active, ce qui complique l'estimation du risque. Dans l'état actuel des connaissances, la possibilité d'un risque de développer des lymphomes, des leucémies ou d'autres maladies malignes chez les patients traités par anti-TNF ne peut être exclue.
Des tumeurs malignes, dont certaines d'issue fatale, ont été rapportées après la commercialisation chez des enfants, des adolescents et des adultes jeunes (jusqu'à l'âge de 22 ans) traités par des anti-TNF (initiation du traitement avant l'âge de 18 ans), y compris l'adalimumab. La moitié de ces cas environ étaient des lymphomes. Les autres cas correspondaient à d'autres types de tumeurs malignes parmi lesquels des cancers rares généralement associés à un contexte d'immunosuppression. Le risque de développer des tumeurs malignes ne peut être exclu chez l'enfant et l'adolescent traités par anti-TNF.
Au cours de la surveillance post-marketing, de rares cas de lymphomes hépatospléniques à lymphocytes T ont été identifiés chez des patients traités par l'adalimumab. Cette forme rare de lymphome à lymphocytes T a une évolution très agressive et est souvent fatale. Certains de ces lymphomes hépatospléniques à lymphocytes T observés avec l'adalimumab sont survenus chez des adultes jeunes ayant un traitement concomitant par l'azathioprine ou par la 6-mercaptopurine utilisé dans les maladies inflammatoires de l'intestin. Le risque potentiel de l'association de l'azathioprine ou de la 6-mercaptopurine avec l'adalimumab doit être soigneusement pris en considération. Un risque de développement de lymphome hépatosplénique à lymphocytes T chez des patients traités par Hulio ne peut pas être exclu (voir rubrique Effets indésirables).
Il n'existe pas d'études chez des patients avec antécédents de tumeurs malignes ou chez lesquels le traitement par l'adalimumab est poursuivi après le développement d'un cancer. En conséquence, une prudence accrue devra être observée lorsqu'on envisage un traitement de ces patients par l'adalimumab (voir rubrique Effets indésirables).
Tous les patients, notamment ceux ayant des antécédents de traitement immunosuppresseur intense ou atteints de psoriasis et ayant des antécédents de puvathérapie, devront être examinés à la recherche d'un cancer cutané, autre que mélanome, avant et pendant le traitement par Hulio. Des cas de mélanome et de carcinome à cellules de Merkel ont été également rapportés chez les patients traités par anti-TNF y compris l'adalimumab (voir rubrique Effets indésirables).
Dans une étude clinique prospective évaluant l'emploi d'un autre agent anti-TNF, l'infliximab, chez des patients souffrant de broncho-pneumopathie chronique obstructive (BPCO), modérée à sévère, on rapporte plus de cancers, surtout du poumon, de la tête et du cou, parmi les patients traités par infliximab comparativement aux patients du groupe contrôle. Tous les patients avaient des antécédents de tabagisme important. Pour cette raison, des précautions doivent être prises dans l'emploi d'un anti- TNF chez des patients souffrant de BPCO, et aussi chez des patients à risque de cancer à cause d'un tabagisme important.
Sur la base des données actuelles, on ne sait pas si le traitement par l'adalimumab influence le risque de développer une dysplasie ou un cancer du côlon. Tous les patients atteints de rectocolite hémorragique qui présentent un risque élevé de dysplasie ou de cancer du côlon (par exemple, les patients atteints de rectocolite hémorragique ancienne ou de cholangite sclérosante primitive) ou qui ont un antécédent de dysplasie ou de cancer du côlon doivent faire l'objet d'un dépistage régulier à la recherche d'une dysplasie avant le traitement et pendant toute l'évolution de leur maladie. Cette évaluation doit inclure une coloscopie et des biopsies conformément aux recommandations locales.
Réactions hématologiques
De rares cas de pancytopénie, y compris d'anémie aplasique, ont été rapportés avec les anti-TNF. Des effets indésirables du système sanguin comprenant des cytopénies médicalement significatives (par ex : thrombocytopénie, leucopénie) ont été observés avec l'adalimumab. Il doit être conseillé à tous les patients de demander immédiatement un avis médical s'ils ont des signes ou des symptômes suggérant des troubles sanguins (par ex : fièvre persistante, contusions, saignements, pâleur) sous Hulio. L'arrêt du traitement par adalimumab devra être envisagé pour les patients chez qui des anomalies sanguines significatives seront confirmées.
Vaccinations
Des réponses anticorps similaires au vaccin pneumococcique valence 23 standard et à la vaccination contre le virus trivalent de la grippe ont été observées dans une étude chez 226 adultes souffrant de polyarthrite rhumatoïde traités par l'adalimumab ou un placebo. Aucune donnée n'est disponible sur la transmission secondaire d'infection par des vaccins vivants chez les patients recevant l'adalimumab.
Chez les enfants et les adolescents, il est recommandé, si possible, que toutes les vaccinations soient à jour conformément aux recommandations vaccinales en vigueur avant l'instauration du traitement par l'adalimumab.
Les patients sous adalimumab peuvent recevoir plusieurs vaccins simultanément, excepté en ce qui concerne des vaccins vivants. L'administration de vaccins vivants (par ex., vaccin BCG) à des nourrissons qui ont été exposés à l'adalimumab in utero n'est pas recommandée pendant les 5 mois suivant la dernière injection d'adalimumab chez la mère pendant la grossesse.
Insuffisance cardiaque congestive
Dans un essai clinique mené avec un autre antagoniste du TNF, on a observé une aggravation de l'insuffisance cardiaque congestive et une augmentation de la mortalité par insuffisance cardiaque congestive. Des cas d'aggravation d'insuffisance cardiaque congestive ont aussi été rapportés chez des patients sous adalimumab. L'adalimumab doit être utilisé avec prudence chez les patients atteints d'insuffisance cardiaque légère (NYHA classes I/II). L'adalimumab est contre-indiqué dans l'insuffisance cardiaque modérée à sévère (voir rubrique Contre-indications). Le traitement par Hulio doit être arrêté chez les patients présentant de nouveaux symptômes ou une aggravation de leurs symptômes d'insuffisance cardiaque congestive.
Processus auto-immuns
Le traitement par Hulio peut entraîner la formation d'anticorps auto-immuns. L'impact d'un traitement à long terme par l'adalimumab sur le développement de maladies auto-immunes est inconnu. Si un patient développe des symptômes évoquant un syndrome de type lupus à la suite d'un traitement par Hulio et présente une réaction positive anti-ADN double brin, le traitement par Hulio ne devra pas être poursuivi (voir rubrique Effets indésirables).
Administration simultanée de traitements de fond (DMARD) biologiques ou d'anti-TNF
Des infections graves ont été observées dans des études cliniques lors de l'administration simultanée d'anakinra et d'un autre anti-TNF, l'étanercept, sans bénéfice clinique supplémentaire comparé à l'étanercept seul. En raison de la nature des effets indésirables observés avec le traitement par l'association étanercept et anakinra, des effets néfastes similaires peuvent aussi résulter de l'association d'anakinra et d'autres anti-TNF. Par conséquent, l'association d'adalimumab et d'anakinra n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
L'administration concomitante d'adalimumab avec d'autres médicaments anti-rhumatismaux de fond biologiques (par exemple anakinra et abatacept) ou avec d'autres anti-TNF n'est pas recommandée en raison de l'augmentation possible du risque d'infections, y compris d'infections graves, et d'autres interactions pharmacologiques potentielles (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Chirurgie
L'expérience concernant la tolérance au cours d'interventions chirurgicales chez les patients traités par l'adalimumab est limitée. La longue demi-vie de l'adalimumab doit être prise en compte si une intervention chirurgicale est prévue. Un patient traité par Hulio nécessitant une intervention chirurgicale doit être surveillé attentivement afin de dépister des infections, et des actions appropriées doivent être entreprises. L'expérience concernant la tolérance de l'adalimumab chez les patients opérés pour arthroplastie est limitée.
Occlusion du grêle
Dans la maladie de Crohn, l'échec au traitement peut indiquer la présence de sténoses fibreuses fixes pouvant nécessiter un traitement chirurgical. Les données disponibles suggèrent que l'adalimumab n'aggrave pas ou ne provoque pas de sténoses.
Sujets âgés
La fréquence des infections graves chez les sujets traités par l'adalimumab âgés de plus de 65 ans (3,7%) est plus élevée que chez les patients de moins de 65 ans (1,5%). Certains cas ont eu une issue fatale. Une attention particulière concernant le risque d'infection doit être apportée lors du traitement des sujets âgés.
Population pédiatrique
Voir
vaccinations ci-dessus.
Excipients à effet notoire
Sorbitol
Ce médicament
contient du sorbitol (E420). Ce médicament ne doit pas être administré aux
patients présentant une intolérance héréditaire au fructose.
Sodium
Ce médicament
contient moins de 1 mmol (23 mg) de sodium par dose
de 0,8 mL, c'est-à-dire qu'il est essentiellement «
sans sodium ».
Résumé du profil de tolérance
L'adalimumab a été étudié chez 9 506 patients dans des essais pivots contrôlés et en ouvert d'une durée de 60 mois et plus. Ces essais ont inclus des patients atteints de polyarthrite rhumatoïde récente ou ancienne, d'arthrite juvénile idiopathique (arthrite juvénile idiopathique polyarticulaire et arthrite liée à l'enthésite) ou des patients souffrant de spondylarthrite axiale (spondylarthrite ankylosante et spondylarthrite axiale sans signes radiographiques de SA), de rhumatisme psoriasique, de la maladie de Crohn, de rectocolite hémorragique, de psoriasis, d'hidrosadénite suppurée et d'uvéite. Les études contrôlées pivots portaient sur 6 089 patients ayant reçu de l'adalimumab et 3 801 patients ayant reçu un placebo ou un comparateur actif pendant la phase contrôlée.
Le pourcentage de patients ayant interrompu le traitement en raison d'effets indésirables pendant la phase en double aveugle, contrôlée, des études pivots a été de 5,9% chez les patients traités par l'adalimumab et de 5,4% chez les patients du groupe contrôle.
Les effets indésirables les plus fréquemment rapportés sont les infections (telles que les rhinopharyngites, les infections des voies respiratoires hautes et les sinusites), les réactions au site d'injection (érythème, démangeaisons, hémorragie, douleur ou gonflement), les céphalées et les douleurs musculo-squelettiques.
Des effets indésirables graves ont été rapportés avec l'adalimumab. Les antagonistes du TNF, tels que l'adalimumab affectent le système immunitaire et leur utilisation peut avoir des répercussions sur les défenses du corps contre les infections et le cancer.
Des infections menaçant le pronostic vital et d'issue fatale (comprenant sepsis, infections opportunistes et tuberculose), des réactivations d'hépatite B et différents cancers (y compris, leucémie, lymphome et lymphome hépatosplénique à lymphocytes T) ont également été rapportés avec l'utilisation de l'adalimumab.
Des effets hématologiques, neurologiques et autoimmuns sévères ont également été rapportés. Ceci comprend de rares cas de pancytopénie, d'anémie médullaire, des cas de démyélinisation centrale et périphérique et des cas de lupus, d'événements liés au lupus et de syndrome de Stevens-Johnson.
Population pédiatrique
En général, la fréquence et le type des événements indésirables observés chez l'enfant et l'adolescent ont été comparables à ceux observés chez les patients adultes.
Liste des effets indésirables
La liste des effets indésirables est basée sur les études cliniques et sur l'expérience après commercialisation et est présentée par classe de systèmes d'organes (SOC) et par fréquence dans le tableau 7 ci-dessous : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité. La fréquence la plus élevée observée dans les diverses indications a été incluse. La présence d'un astérisque (*) dans la colonne « Classe de systèmes d'organes » indique que de plus amples informations sont disponibles aux rubriques Contre-indications, Mises en garde spéciales et précautions d'emploi et Effets indésirables.
Tableau 7 : Effets indésirables
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Infections et infestations* | Très fréquent | Infection des voies respiratoires (y compris infection des voies respiratoires supérieures et inférieures, pneumonie, sinusite, pharyngite, rhinopharyngite et pneumonie herpétique) |
| Fréquent |
Infections systémiques (y compris
sepsis, candidose et grippe). Infections intestinales (y compris
gastro-entérite virale). Infections cutanées et des tissus mous (y compris panaris superficiel périunguéal, cellulite, impétigo, fasciite nécrosante et zona). Infections de l'oreille. Infections buccales (y compris herpès, herpès buccal et infections dentaires). Infections des organes de reproduction (y compris mycose vulvo-vaginale). Infections des voies urinaires (y compris pyélonéphrite). Infections fongiques. Infections articulaires. |
|
| Peu fréquent |
Infections neurologiques (y
compris méningite virale). Infections opportunistes et tuberculose (y compris coccidioïdomycose, histoplasmose et infection à Mycobacterium avium complex). Infections bactériennes. Infections oculaires. Diverticulite1) |
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Tumeurs bénignes, malignes et non précisées (y compris kystes et polypes)* | Fréquent |
Cancer de la peau, à l'exclusion
du mélanome (y compris carcinome basocellulaire et
carcinome spino-cellulaire). Tumeur bénigne. |
| Peu fréquent |
Lymphome**. Tumeur des organes solides (y compris cancer du sein, tumeur du poumon et tumeur de la thyroïde). Mélanome**. |
|
| Rare | Leucémie1). | |
| Fréquence indéterminée |
Lymphome hépatosplénique à
lymphocytes T1), Carcinome à cellules de Merkel (carcinome
neuroendocrine cutané)1). Sarcome de Kaposi. |
|
| Affections hématologiques et du système lymphatique* | Très fréquent |
Leucopénie (y compris neutropénie
et agranulocytose). Anémie. |
| Fréquent |
Leucocytose. Thrombocytopénie. |
|
| Peu fréquent | Purpura thrombopénique idiopathique | |
| Rare | Pancytopénie | |
| Affections du système immunitaire* | Fréquent |
Hypersensibilité. Allergies (y compris allergie saisonnière). |
| Peu fréquent |
Sarcoïdose1). Vasculite. |
|
| Rare | Anaphylaxie1). | |
| Troubles du métabolisme et de la nutrition | Très fréquent | Augmentation du taux de lipides. |
| Fréquent |
Hypokaliémie. Augmentation de l'acide urique. Taux anormal de sodium dans le sang. Hypocalcémie. Hyperglycémie. Hypophosphatémie. Déshydratation. |
|
| Affections psychiatriques | Fréquent |
Troubles de l'humeur (y compris
dépression). Anxiété. Insomnie. |
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Affections du système nerveux* | Très fréquent | Céphalées. |
| Fréquent |
Paresthésies (y compris
hypoesthésie). Migraine. Compression des racines nerveuses. |
|
| Peu fréquent |
Accident vasculaire cérébral1).
Tremblements. Neuropathie. |
|
| Rare |
Sclérose en plaques. Affections démyélinisantes (par ex. névrite optique, syndrome de Guillain-Barré)1). |
|
| Affections oculaires | Fréquent |
Troubles visuels. Conjonctivite. Blépharite. Gonflement des yeux. |
| Peu fréquent | Diplopie | |
| Affections de l'oreille et du labyrinthe | Fréquent | Vertige |
| Peu fréquent |
Surdité. Acouphènes. |
|
| Affections cardiaques* | Fréquent | Tachycardie |
| Peu fréquent |
Infarctus du myocarde1).
Arythmies. Insuffisance cardiaque congestive. |
|
| Rare | Arrêt cardiaque | |
| Affections vasculaires | Fréquent |
Hypertension. Bouffées de chaleur. Hématomes. |
| Peu fréquent |
Anévrisme aortique. Occlusion vasculaire. Thrombophlébite. |
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Affections respiratoires, thoraciques et médiastinales* | Fréquent |
Asthme. Dyspnée. Toux. |
| Peu fréquent |
Embolie pulmonaire1). Maladie pulmonaire interstitielle. Broncho-pneumopathie chronique obstructive. Pneumopathie. Épanchement pleural1) |
|
| Rare | Fibrose pulmonaire1) | |
| Affections gastro- intestinales | Très fréquent |
Douleurs abdominales, Nausées et vomissements. |
| Fréquent |
Hémorragie gastro-intestinale. Dyspepsie. Reflux gastro-œsophagien. Syndrome de Gougerot-Sjögren. |
|
| Peu fréquent |
Pancréatite. Dysphagie. Œdème du visage. |
|
| Rare | Perforation intestinale1) | |
| Affections hépatobiliaires* | Très fréquent | Enzymes hépatiques élevées |
| Peu fréquent |
Cholécystites et lithiase
biliaire. Stéatose hépatique. Hyperbilirubinémie. |
|
| Rare |
Hépatite. Réactivation d'hépatite B1). Hépatite auto-immune1). |
|
| Fréquence indéterminée | Insuffisance hépatique1) |
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Affections de la peau et du tissu sous-cutané | Très fréquent | Rash (y compris éruption exfoliative). |
| Fréquent |
Aggravation ou apparition d'un
psoriasis (y compris psoriasis pustulaire palmoplantaire)1). Urticaire. Contusions (y compris purpura). Dermatite (y compris eczéma). Onychoclasie. Hyperhidrose. Alopécie1). Prurit |
|
| Peu fréquent |
Sueurs nocturnes. Cicatrice. |
|
| Rare |
Érythème polymorphe1). Syndrome de Stevens-Johnson1). Angiœdème1). Vascularite cutanée1). Réaction lichénoïde cutanée1). |
|
| Fréquence indéterminée | Aggravation des symptômes de dermatomyosite1). | |
| Affections musculo- squelettiques et systémiques | Très fréquent | Douleurs musculo-squelettiques. |
| Fréquent | Spasmes musculaires (y compris augmentation de la créatine phosphokinase sérique). | |
| Peu fréquent |
Rhabdomyolyse. Lupus érythémateux disséminé. |
|
| Rare | Syndrome de type lupus1). | |
| Affections du rein et des voies urinaires | Fréquent |
Insuffisance rénale. Hématurie. |
| Peu fréquent | Nycturie | |
| Affections des organes de reproduction et du sein | Peu fréquent | Troubles de la fonction érectile. |
| Troubles généraux et anomalies au site d'administration* | Très fréquent | Réaction au site d'injection (y compris érythème au site d'injection) |
| Fréquent |
Douleur thoracique. Œdème. Fièvre1). |
|
| Peu fréquent | Inflammation |
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Investigations* | Fréquent |
Troubles de la coagulation et
troubles hémorragiques (y compris allongement du temps de céphaline activée).
Positivité aux auto-anticorps (y compris aux anticorps anti-ADN double brin). Augmentation du taux sanguin de lactate déshydrogénase. |
| Fréquence indéterminée | Augmentation du poids2) | |
| Lésions, intoxications et complications liées aux procédures | Fréquent | Retard de cicatrisation |
** y compris les études d'extension en ouvert.
1) y compris les données des notifications spontanées
2) Le changement de poids moyen par rapport aux valeurs initiales pour l'adalimumab allait de 0,3 kg à 1,0 kg pour toutes les indications chez l'adulte, contre (moins) -0,4 kg à 0,4 kg pour le placebo, sur une période de traitement de 4 à 6 mois. Une augmentation de poids comprise entre 5 et 6 kg a également été observée au cours d'études d'extension à long terme, avec des expositions moyennes d'environ 1 à 2 ans sans groupe témoin, en particulier chez les patients atteints de la maladie de Crohn et de colite ulcéreuse. Le mécanisme qui sous-tend cet effet n'est pas clair mais pourrait être associé à l'action anti-inflammatoire de l'adalimumab.
Hidrosadénite suppurée
Le profil de sécurité chez les patients atteints d'hidrosadénite suppurée traités par l'adalimumab de façon hebdomadaire correspond au profil de sécurité connu de l'adalimumab.
Uvéite
Le profil de sécurité chez les patients atteints d'uvéite traités par l'adalimumab toutes les deux semaines correspond au profil de sécurité connu de l'adalimumab.
Description des effets indésirables sélectionnés
Réactions au site d'injection
Dans les essais contrôlés pivots menés chez l'adulte et l'enfant, 12,9% des patients traités par l'adalimumab ont présenté des réactions au site d'injection (érythème et/ou prurit, saignement, douleur ou tuméfaction) contre 7,2% des patients recevant le placebo ou le comparateur actif. Les réactions au point d'injection n'ont généralement pas nécessité l'arrêt du médicament.
Infections
Dans les essais contrôlés pivots menés chez l'adulte et l'enfant, la fréquence des infections a été de 1,51 par patient-année dans le groupe de l'adalimumab, et de 1,46 par patient-année dans le groupe placebo et le groupe contrôle. Les infections consistaient essentiellement en rhinopharyngites, infections de l'appareil respiratoire supérieur et sinusites. La plupart des patients ont continué l'adalimumab après la guérison de l'infection.
L'incidence des infections graves a été de 0,04 par patient-année dans le groupe de l'adalimumab, et de 0,03 par patient-année dans le groupe placebo et le groupe contrôle.
Dans les études contrôlées et en ouvert avec l'adalimumab menées chez l'adulte et dans la population pédiatrique, des infections graves (y compris des infections d'issue fatale, ce qui s'est produit rarement) ont été rapportées dont des signalements de tuberculose (y compris miliaire et à localisations extra-pulmonaires) et d'infections opportunistes invasives (par ex. histoplasmose disséminée ou histoplasmose extrapulmonaire, blastomycose, coccidioïdomycose, pneumocystose, candidose, aspergillose et listériose). La plupart des cas de tuberculose sont survenus dans les huit premiers mois après le début du traitement et peuvent être le reflet d'une réactivation d'une maladie latente.
Tumeurs malignes et troubles lymphoprolifératifs
Aucun cas de cancer n'a été observé chez 249 patients pédiatriques représentant une exposition de 655,6 patient-années lors des études avec l'adalimumab chez les patients atteints d'arthrite juvénile idiopathique (arthrite juvénile idiopathique polyarticulaire et arthrite liée à l'enthésite). De plus, aucun cas de cancer n'a été observé chez 192 patients pédiatriques représentant une exposition de 498,1 patient-années lors des études avec l'adalimumab dans la maladie de Crohn chez l'enfant et l'adolescent. Aucun cas de cancer n'a été observé chez 77 patients pédiatriques correspondant à une exposition de 80 patient-années lors d'une étude avec l'adalimumab dans le psoriasis en plaques chronique pédiatrique. Lors d'une étude avec l'adalimumab menée chez des enfants et des adolescents atteints de rectocolite hémorragique, aucun cas de cancer n'a été observé chez 93 enfants et adolescents représentant une exposition de 65,3 patient-années. Aucun cas de cancer n'a été observé chez 60 patients pédiatriques représentant une exposition de 58,4 patient-années lors d'une étude avec l'adalimumab dans l'uvéite pédiatrique.
Pendant les
périodes contrôlées des essais cliniques pivots chez l'adulte avec l'adalimumab d'une durée d'au moins 12 semaines chez des
patients souffrant de polyarthrite rhumatoïde modérément à sévèrement active,
de spondylarthrite ankylosante, de spondylarthrite axiale sans signes
radiographiques de SA, de rhumatisme psoriasique, de psoriasis, d'hidrosadénite
suppurée, de la maladie de Crohn, de rectocolite
hémorragique et d'uvéite, un taux (intervalle de confiance à 95%) de cancers
autres que lymphomes ou cancers de la peau non mélanomes, de 6,8 (4,4 - 10,5)
pour1 000 patient-années parmi les 5 291 patients traités par l'adalimumab, a été observé par rapport à un taux de
6,3 (3,4 - 11,8) pour 1 000 patient-années parmi les 3 444 patients du groupe
contrôle (la durée moyenne du traitement était de 4,0 mois pour les patients
traités par l'adalimumab et de 3,8 mois pour les
patients du groupe contrôle). Le taux (intervalle de confiance à 95%) de
cancers de la peau non mélanomes était de 8,8 (6,0 - 13,0) pour 1 000
patient-années pour les patients traités par l'adalimumab
et de 3,2 (1,3 - 7,6) pour 1 000 patient-années parmi les patients du groupe
contrôle.
Dans ces cancers
de la peau, les carcinomes spino-cellulaires sont
survenus à des taux (intervalle de confiance à 95%) de 2,7 (1,4 - 5,4) pour 1
000 patient-années chez les patients traités par l'adalimumab
et 0,6 (0,1 - 4,5) pour 1 000 patient-années chez les patients du groupe
contrôle. Le taux (intervalle de confiance à 95%) de lymphomes était de 0,7
(0,2 - 2,7) pour 1 000 patient-années chez les patients traités par l'adalimumab et 0,6 (0,1 - 4,5) pour 1 000 patient-années
chez les patients du groupe contrôle.
En joignant les périodes contrôlées de ces essais et les essais d'extension en ouvert terminés ou en cours avec une durée moyenne d'environ 3,3 ans incluant 6 427 patients et plus de 26 439 patient- années de traitement, le taux observé de cancers, autres que lymphomes et cancers de la peau non mélanomes est d'environ 8,5 pour 1 000 patient-années. Le taux observé de cancers de la peau non- mélanomes est d'environ 9,6 pour 1 000 patient-années et le taux de lymphomes observés est d'environ 1,3 pour 1 000 patient-années.
En post-marketing de janvier 2003 à décembre 2010, essentiellement chez les patients atteints de polyarthrite rhumatoïde, le taux rapporté de cancers est approximativement de 2,7 pour 1 000 patient- années de traitement. Les taux rapportés pour les cancers de la peau non-mélanomes et les lymphomes sont respectivement d'environ 0,2 et 0,3 pour 1 000 patient-années de traitement (voir rubrique Mises en garde spéciales et précautions d'emploi).
Au cours de la surveillance post-marketing, de rares cas de lymphome hépatosplénique à lymphocytes T ont été rapportés chez des patients traités par l'adalimumab (voir rubrique Mises en garde spéciales et précautions d'emploi).
Auto-anticorps
Des recherches d'auto-anticorps répétées ont été effectuées sur des échantillons de sérum des patients des essais I - V dans la polyarthrite rhumatoïde. Dans ces essais, les titres d'anticorps antinucléaires initialement négatifs étaient positifs à la semaine 24 chez 11,9% des patients traités par l'adalimumab et 8,1% des patients sous placebo et comparateur. Deux patients sur les 3 441 traités par l'adalimumab dans toutes les études dans la polyarthrite rhumatoïde et le rhumatisme psoriasique ont présenté des signes cliniques évoquant un syndrome pseudo-lupique d'apparition nouvelle. L'état des patients s'est amélioré après l'arrêt du traitement. Aucun patient n'a présenté de néphrite lupique ou de symptômes nerveux centraux.
Événements hépatobiliaires
Dans les essais cliniques contrôlés de phase III portant sur l'adalimumab dans la polyarthrite rhumatoïde et le rhumatisme psoriasique avec une période de contrôle de 4 à 104 semaines, des élévations d'ALAT ≥ 3 × LSN sont survenues chez 3,7% des patients traités par l'adalimumab et chez 1,6% des patients du groupe contrôle.
Dans les
essais cliniques contrôlés de phase III portant sur l'adalimumab
chez les patients atteints d'arthrite juvénile idiopathique polyarticulaire
âgés de 4 à 17 ans et les patients atteints d'arthrite liée à l'enthésite âgés de 6 à 17 ans, des élévations d'ALAT ≥
3 × LSN sont survenues chez 6,1% des patients traités par l'adalimumab
et chez 1,3% des patients du groupe contrôle. La plupart des élévations d'ALAT
sont survenues dans le cadre d'une utilisation concomitante de méthotrexate.
Aucune
élévation d'ALAT ≥ 3 × LSN n'est survenue au cours de l'essai de phase
III de l'adalimumab chez des patients atteints
d'arthrite juvénile idiopathique polyarticulaire âgés
de 2 ans à moinsde 4 ans.
Dans les essais cliniques contrôlés de phase III portant sur l'adalimumab chez les patients atteints de la maladie de Crohn et de rectocolite hémorragique avec une période de contrôle de 4 à 52 semaines, des élévations d'ALAT ≥ 3 × LSN sont survenues chez 0,9% des patients traités par l'adalimumab et chez 0,9% des patients du groupe contrôle.
Dans l'essai clinique de phase III portant sur l'adalimumab chez les enfants et adolescents atteints de la maladie de Crohn qui a évalué l'efficacité et le profil de sécurité de deux schémas posologiques d'entretien en fonction du poids après un traitement d'induction ajusté au poids jusqu'à 52 semaines de traitement, des élévations d'ALAT ≥ 3 × LSN sont survenues chez 2,6% des patients (5/192), parmi lesquels 4 étaient traités en association avec des immunosuppresseurs au début de l'étude.
Dans les essais cliniques contrôlés de phase III portant sur l'adalimumab dans le psoriasis en plaques avec une période de contrôle de 12 à 24 semaines, des élévations d'ALAT ≥ 3 × LSN sont survenues chez 1,8% des patients traités par l'adalimumab et chez 1,8% des patients du groupe contrôle.
Il n'a pas été observé d'élévations de l'ALAT ≥ 3 × LSN dans l'étude de phase III portant sur l'adalimumab chez des patients pédiatriques atteints de psoriasis en plaques.
Dans les essais cliniques contrôlés portant sur l'adalimumab (doses initiales de 160 mg à la semaine 0 et 80 mg à la semaine 2, suivies de 40 mg chaque semaine à partir de la semaine 4), chez les patients atteints d'hidrosadénite suppurée avec une période de contrôle de 12 à 16 semaines, des élévations d'ALAT ≥ 3 × LSN sont survenues chez 0,3% des patients traités par l'adalimumab et 0,6% des patients du groupe contrôle.
Dans les essais cliniques contrôlés portant sur l'adalimumab (dose initiale de 80 mg à la semaine 0 suivie de 40 mg toutes les deux semaines à partir de la semaine 1) chez les patients adultes atteints d'uvéite pour une durée allant jusqu'à 80 semaines, avec une durée médiane d'exposition de respectivement 166,5 jours et 105,0 jours pour les patients traités par l'adalimumab et les patients du groupe contrôle, des élévations d'ALAT ≥ 3 × LSN sont survenues chez 2,4% des patients traités par l'adalimumab et 2,4% des patients du groupe contrôle.
Dans l'essai clinique contrôlé de phase III de l'adalimumab mené chez des enfants et des adolescents atteints de rectocolite hémorragique (N = 93) qui a évalué l'efficacité et le profil de sécurité d'une dose d'entretien de 0,6 mg/kg (dose maximale de 40 mg) administrée une semaine sur deux (N = 31) et d'une dose d'entretien de 0,6 mg/kg (dose maximale de 40 mg) administrée chaque semaine (N = 32), à la suite d'une dose d'induction ajustée en fonction du poids corporel de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0 et à la semaine 1, et de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 (N = 63), ou d'une dose d'induction de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0, d'un placebo à la semaine 1, et d'une dose de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 (N = 30), des élévations de l'ALAT ≥ 3 x LSN sont survenues chez 1,1 % (1/93) des patients.
Dans les essais cliniques, toutes indications confondues, les patients avec ALAT augmentées étaient asymptomatiques et dans la plupart des cas les élévations étaient transitoires et réversibles lors de la poursuite du traitement. Cependant, au cours de la surveillance post-marketing, des insuffisances hépatiques ainsi que des désordres hépatiques moins sévères, qui peuvent précéder une insuffisance hépatique, tels que des hépatites y compris des hépatites auto-immunes, ont été rapportés chez des patients recevant de l'adalimumab.
Administration concomitante d'azathioprine/6-mercaptopurine
Lors d'études dans la maladie de Crohn chez l'adulte, une incidence plus élevée de tumeurs et d'infections graves a été observée avec l'association adalimumab et azathioprine/6-mercaptopurine comparativement à l'adalimumab utilisé seul.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT L'INSTAURATION DU TRAITEMENT, recherche d'infection tuberculeuse active ou non (latente) avec :
-
évaluation médicale détaillée chez les patients ayant des antécédents
de tuberculose ou d'exposition antérieure possible à des patients
atteints de tuberculose active et/ou d'un traitement immunosuppresseur
actuel ou ancien;
- tests de dépistage appropriés, par exemple test
dermique à la tuberculine et radiographie pulmonaire. Il est conseillé
de noter la réalisation et les résultats de ces tests dans la carte de
surveillance du patient. Il est rappelé aux prescripteurs que le test
dermique à la tuberculine peut donner des faux-négatifs notamment chez
les patients gravement malades ou immunodéprimés;
- envisage une prophylaxie antituberculeuse avant l'instauration du
traitement chez les patients ayant des facteurs de risque multiples ou
significatifs de tuberculose malgré un test de dépistage de la
tuberculose négatif et chez les patients ayant des antécédents de
tuberculose latente ou active, chez qui l'administration d'un
traitement anti-tuberculeux approprié ne peut être confirmée;
- dépistage d'infection à VHB;
- évaluation neurologique chez les patients présentant une uvéite intermédiaire non infectieuse;
- s'assurer que les vaccinations soient à jour conformément aux recommandations vaccinales en vigueur chez les enfants et les adolescents.
SURVEILLANCE pendant le traitement :
- infections, jusqu'à 4 mois après l'arrêt du traitement.
- évaluation
neurologique chez les patients présentant une uvéite
intermédiaire non infectieuse régulièrement au cours du traitement;
- examen à la recherche d'un cancer cutané autre que mélanome.
L'administration de vaccins vivants (par exemple, vaccin BCG) à des
enfants qui ont été exposés à l'adalimumab in utero n'est pas
recommandée pendant les 5 mois suivant la dernière injection de la mère
durant la grossesse.
ARRETER LE TRAITEMENT ET CONTACTER IMMEDIATEMENT LE MEDECIN en cas de :
- éruption cutanée sévère, urticaire;
- gonflement de la face, des mains, des pieds;
- gêne respiratoire, gêne en avalant;
- essoufflement au cours de l'activité physique ou en position allongée ou gonflement des pieds;
- oppression dans la poitrine, sensations vertigineuses.
INFORMER IMMEDIATEMENT LE MEDECIN en cas de :
- toux persistante, perte de poids, manque d'énergie, légère fièvre;
- modifications de la vision (vision double), faiblesse dans les bras
ou les jambes, engourdissement ou fourmillements dans une partie du
corps;
- fièvre persistante, contusions, saignements, pâleur.
INFORMER LE MEDECIN en cas de :
- fièvre, plaies, sensations de nausées ou de malaise, problèmes dentaires, brûlures en urinant;
- sensation de faiblesse ou de fatigue;
- intervention chirurgicale ou dentaire programmée;
- nouvelles lésions cutanées apparaissant pendant le traitement ou changement d'aspect de lésions pré-existantes;
- rash persistant inexpliqué, fièvre, douleur articulaire ou fatigue.
FEMME EN AGE DE PROCREER :
- Utiliser une méthode de contraception
efficace pendant le traitement et pendant au moins 5 mois après l'arrêt
du traitement.
- En cas de prise de ce traitement pendant la grossesse,
INFORMER LE MEDECIN du bébé.
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (vertiges, troubles visuels).
Femmes en âge de procréer
Les femmes en âge de procréer doivent envisager l'utilisation d'une méthode de contraception efficace pour éviter toute grossesse et la poursuivre pendant cinq mois au moins après la dernière administration d'Hulio.
Grossesse
Un grand nombre (environ 2 100) de grossesses exposées à l'adalimumab dont les données ont été recueillies prospectivement, aboutissant à une naissance vivante avec une évolution à terme connue, avec notamment plus de 1 500 grossesses exposées à l'adalimumab au cours du premier trimestre, ne révèle aucune augmentation du taux de malformations chez le nouveau-né.
Au
cours d'une étude de cohorte prospective, 257 femmes présentant une
polyarthrite rhumatoïde (PR) ou une maladie de Crohn (MC) et traitées
par l'adalimumab au moins pendant le premier trimestre et 120 femmes
présentant une PR ou une MC non traitées par l'adalimumab ont été
incluses. La prévalence à la naissance des anomalies congénitales
majeures constituait le critère d'évaluation principal. Le taux de
grossesses aboutissant à au moins un nouveau-né en vie présentant une
anomalie congénitale majeure était de 6/69 (8,7%) chez les femmes
traitées par l'adalimumab présentant une PR et de 5/74 (6,8%) chez les
femmes non traitées présentant une PR (OR non ajusté 1,31, IC à 95%
0,38- 4,52), et de 16/152 (10,5%) chez les femmes traitées par
l'adalimumab présentant une MC et de 3/32 (9,4%) chez les femmes non
traitées présentant une MC (OR non ajusté 1,14, IC à 95% 0,31-4,16).
L'OR ajusté (compte tenu des différences initiales) était de 1,10 (IC à
95% 0,45-2,73) pour les PR et MC combinées. Aucune différence notable
n'a été rapportée entre les femmes traitées par l'adalimumab et les
femmes non traitées par l'adalimumab pour les critères d'évaluation
secondaires d'avortements spontanés, d'anomalies congénitales mineures,
d'accouchement prématuré, de taille à la naissance et d'infections
graves ou opportunistes, et de mortinatalité ou de malignité.
L'interprétation des données peut être affectée en raison des limites
méthodologiques de l'étude, notamment la petite taille d'échantillon et
le plan d'étude non randomisé.
Dans
une étude de toxicité sur le développement réalisée chez des singes, il
n'y a eu aucun signe évocateur d'une éventuelle toxicité maternelle,
d'embryo-toxicité ou de potentiel tératogène.
On ne dispose pas de données précliniques sur la toxicité post-natale de l'adalimumab (voir rubrique Données de sécurité préclinique).
En raison de son effet inhibiteur sur le TNFα, l'adalimumab administré pendant la grossesse pourrait affecter les réponses immunitaires normales du nouveau-né. L'adalimumab doit être utilisé pendant la grossesse seulement si nécessaire.
Chez les femmes traitées par l'adalimumab durant leur grossesse, l'adalimumab peut traverser le placenta et passer dans le sang de leur enfant. En conséquence, ces enfants peuvent avoir un risque accru d'infections. L'administration de vaccins vivants (par exemple, vaccin BCG) à des enfants qui ont été exposés à l'adalimumab in utero n'est pas recommandée pendant les 5 mois suivant la dernière injection de la mère durant la grossesse.
Allaitement
Des données limitées issues de la littérature publiée indiquent que l'adalimumab est excrété dans le lait maternel à de très faibles concentrations, l'adalimumab étant présent dans le lait maternel à des concentrations équivalentes à 0,1%-1% des taux sériques maternels. Administrées par voie orale, les protéines immunoglobulines G subissent une protéolyse intestinale et présentent une faible biodisponibilité. Aucun effet sur les nouveau-nés/nourrissons allaités n'est attendu. Par conséquent, l'adalimumab peut être utilisé pendant l'allaitement.
Fertilité
On ne dispose pas de données précliniques sur les effets de l'adalimumab sur la fertilité.
L'adalimumab a été étudié chez des patients atteints de polyarthrite rhumatoïde, d'arthrite juvénile idiopathique polyarticulaire et de rhumatisme psoriasique prenant de l'adalimumab en monothérapie et chez ceux prenant simultanément du méthotrexate. La formation d'anticorps était plus faible lorsque l'adalimumab était administré en même temps que du méthotrexate par comparaison avec son utilisation en monothérapie. L'administration de l'adalimumab sans méthotrexate a entraîné une augmentation de la formation d'anticorps, une augmentation de la clairance et une réduction de l'efficacité de l'adalimumab (voir rubrique Propriétés pharmacodynamiques).
L'association d'Hulio et d'anakinra n'est pas recommandée (voir rubrique Mises en garde spéciales et précautions d'emploi « Administration simultanée de traitements de fond (DMARD) biologiques et d'anti-TNF »).
L'association d'Hulio et d'abatacept n'est pas recommandée (voir rubrique Mises en garde spéciales et précautions d'emploi « Administration simultanée de traitements de fond (DMARD) biologiques et d'anti-TNF »).
Le traitement par Hulio doit être instauré et supervisé par un médecin spécialiste qualifié en matière de diagnostic et de traitement des pathologies dans lesquelles Hulio est indiqué. Il est recommandé aux ophtalmologistes de consulter un spécialiste approprié avant d'instaurer un traitement par Hulio (voir rubrique Mises en garde spéciales et précautions d'emploi). Une carte de surveillance sera remise aux patients traités par Hulio.
Après une formation correcte à la technique d'injection, les patients peuvent s'auto-injecter Hulio, si leur médecin l'estime possible, et à condition d'un suivi médical approprié.
Pendant le traitement par Hulio, les autres traitements concomitants (tels que les corticoïdes et/ou immunomodulateurs) devront être optimisés.
Posologie
Population pédiatrique
Arthrite juvénile idiopathique
Arthrite juvénile idiopathique polyarticulaire à partir de 2 ans
La posologie recommandée d'Hulio pour les patients atteints d'arthrite juvénile idiopathique polyarticulaire à partir de l'âge de 2 ans dépend du poids corporel (tableau 1). Hulio est administré toutes les deux semaines, en injection sous-cutanée.
Tableau 1 : Posologie d'Hulio chez les patients atteints d'arthrite juvénile idiopathique polyarticulaire
| Poids du patient | Schéma posologique |
| 10 kg à < 30 kg | 20 mg toutes les deux semaines |
| ≥30 kg | 40 mg toutes les deux semaines |
Les données disponibles laissent supposer que la réponse clinique est habituellement obtenue en 12 semaines de traitement. La poursuite du traitement devra être soigneusement reconsidérée chez un patient n'ayant pas répondu dans ces délais.
Il n'y a pas d'utilisation justifiée d'adalimumab chez les patients âgés de moins de 2 ans dans cette indication.
D'autres dosages et/ou présentations d'Hulio peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Arthrite liée à l'enthésite
La posologie recommandée d'Hulio pour les patients atteints d'arthrite liée à l'enthésite à partir de l'âge de 6 ans dépend du poids corporel (tableau 2). Hulio est administré toutes les deux semaines, en injection sous-cutanée.
Tableau 2 : Posologie d'Hulio chez les patients atteints d'arthrite liée à l'enthésite
| Poids du patient | Schéma posologique |
| 15 kg à < 30 kg | 20 mg toutes les deux semaines |
| ≥30 kg | 40 mg toutes les deux semaines |
L'adalimumab n'a pas été étudié chez les patients de moins de 6 ans atteints d'arthrite liée à l'enthésite.
D'autres dosages et/ou présentations d'Hulio peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Psoriasis en plaques chez l'enfant et l'adolescent
La posologie recommandée d'Hulio pour les patients atteints de psoriasis en plaques âgés de 4 à 17 ans dépend du poids corporel (tableau 3). Hulio est administré en injection sous-cutanée.
Tableau 3 : Posologie d'Hulio chez les enfants et les adolescents atteints de psoriasis en plaques
| Poids du patient | Schéma posologique |
| 15 kg à < 30 kg | Dose initiale de 20 mg puis de 20 mg toutes les deux semaines en commençant une semaine après l'administration de la première dose |
| ≥30 kg | Dose initiale de 40 mg puis de 40 mg toutes les deux semaines en commençant une semaine après l'administration de la première dose |
La poursuite du traitement au-delà de 16 semaines doit être soigneusement reconsidérée chez un patient n'ayant pas répondu dans ces délais.
Si un retraitement par Hulio est indiqué, les recommandations mentionnées ci-dessus pour la posologie et la durée de traitement doivent être suivies.
La sécurité de l'adalimumab chez l'enfant et l'adolescent présentant un psoriasis en plaques a été évaluée sur une durée moyenne de 13 mois.
Il n'existe pas d'utilisation justifiée de l'adalimumab chez les enfants âgés de moins de 4 ans dans cette indication.
D'autres dosages et/ou présentations d'Hulio peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Hidrosadénite suppurée de l'adolescent (à partir de 12 ans, pesant au moins 30 kg)
Il n'existe pas d'essai clinique conduit avec l'adalimumab chez des adolescents atteints d'hidrosadénite suppurée (HS).
La posologie de l'adalimumab chez ces patients a été déterminée à partir d'une modélisation pharmacocinétique et d'une simulation (voir rubrique Propriétés pharmacocinétiques).
La posologie recommandée d'Hulio est de 80 mg à la semaine 0 suivie de 40 mg toutes les deux semaines à partir de la semaine 1 en injection sous-cutanée.
Chez les adolescents présentant une réponse insuffisante à Hulio 40 mg toutes les deux semaines, une augmentation de la posologie à 40 mg toutes les semaines ou 80 mg toutes les deux semaines peut être envisagée.
Si nécessaire, les antibiotiques peuvent être poursuivis au cours du traitement par Hulio. Au cours du traitement par Hulio, il est recommandé au patient de nettoyer quotidiennement ses lésions avec un antiseptique topique.
La poursuite du traitement au-delà de 12 semaines doit être soigneusement reconsidérée chez les patients ne présentant pas d'amélioration pendant cette période.
Si le traitement est interrompu, Hulio peut être réintroduit si nécessaire.
Le bénéfice et le risque d'un traitement continu à long terme doivent faire l'objet d'une évaluation régulière (voir les données chez les adultes à la rubrique Propriétés pharmacodynamiques).
Il n'existe pas d'utilisation justifiée de l'adalimumab chez les enfants âgés de moins de 12 ans dans cette indication.
D'autres dosages et/ou présentations d'Hulio peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Maladie de Crohn de l'enfant et l'adolescent
La posologie recommandée d'Hulio pour les patients atteints de la maladie de Crohn âgés de 6 à 17 ans dépend du poids corporel (tableau 4). Hulio est administré en injection sous-cutanée.
Tableau 4 : Posologie d'Hulio chez les enfants et les adolescents atteints de la maladie de Crohn
| Poids du patient | Dose d'induction | Dose d'entretien à partir de la semaine 4 |
| < 40 kg |
· 40 mg en
semaine 0 et 20 mg en semaine 2
S'il est nécessaire d'obtenir une
réponse plus rapide au traitement, sachant que le risque d'événements
indésirables peut être plus important avec une dose d'induction plus élevée,
la posologie suivante peut être administrée :
· 80 mg en
semaine 0 et 40 mg en semaine 2
|
20 mg toutes les deux semaines |
| ≥40 kg |
· 80 mg en
semaine 0 et 40 mg en semaine 2
S'il est nécessaire d'obtenir une
réponse plus rapide au traitement, sachant que le risque d'événements
indésirables peut être plus important avec une dose d'induction plus élevée,
la posologie suivante peut être administrée :
· 160 mg en
semaine 0 et 80 mg en semaine 2
|
40 mg toutes les deux semaines |
Les patients chez qui une réponse insuffisante au traitement est observée peuvent bénéficier d'une augmentation de la posologie :
· < 40 kg : 20 mg par semaine
· ≥ 40 kg : 40 mg par semaine ou 80 mg toutes les deux semaines
La poursuite du traitement devra être soigneusement reconsidérée chez un patient n'ayant pas répondu à la semaine 12.
Il n'existe pas d'utilisation justifiée de l'adalimumab chez les enfants âgés de moins de 6 ans dans cette indication.
D'autres dosages et/ou présentations d'Hulio peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Rectocolite hémorragique chez l'enfant et l'adolescent
La posologie recommandée d'Hulio pour les patients âgés de 6 à 17 ans et atteints de rectocolite hémorragique dépend du poids corporel (tableau 5). Hulio est administré par injection sous-cutanée.
Tableau 5 : Posologie d'Hulio chez les enfants et les adolescents atteints de rectocolite hémorragique
| Poids du patient | Dose d'induction | Dose d'entretien à partir de la semaine 4* |
| < 40 kg |
· 80 mg à la
semaine 0 (deux injections de 40 mg le jour de l'induction) et 40 mg à la
semaine 2 (une seule injection de 40 mg)
|
40 mg toutes les deux semaines |
| ≥ 40 kg |
· 160 mg à la
semaine 0 (quatre injections de 40 mg le jour de l'induction ou deux
injections de 40 mg par jour pendant deux jours) et 80 mg à la semaine 2
(deux injections de 40 mg le jour de l'injection)
|
80 mg toutes les deux semaines |
La poursuite du traitement au-delà de 8 semaines doit être soigneusement reconsidérée chez les patients n'ayant pas répondu pendant cette période.
Il n'y a pas d'utilisation justifiée d'Hulio chez les enfants âgés de moins de 6 ans dans cette indication.
Différents dosages et/ou présentations d'Hulio peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Uvéite chez l'enfant et l'adolescent
La posologie recommandée d'Hulio pour les enfants et les adolescents atteints d'uvéite à partir de l'âge de 2 ans dépend du poids corporel (tableau 6). Hulio est administré en injection sous-cutanée.
Dans l'uvéite chez l'enfant et l'adolescent, aucun essai clinique n'a été conduit avec Hulio sans traitement concomitant par le méthotrexate.
Tableau 6 : Posologie d'Hulio chez les enfants et les adolescents atteints d'uvéite
| Poids du patient | Schéma posologique |
| < 30 kg | 20 mg toutes les deux semaines en association avec le méthotrexate |
| ≥30 kg | 40 mg toutes les deux semaines en association avec le méthotrexate |
Lors de l'instauration du traitement par Hulio, une dose de charge de 40 mg pour les patients < 30 kg ou de 80 mg pour les patients ≥ 30 kg peut être administrée une semaine avant le début du traitement d'entretien. Aucune donnée clinique n'est disponible sur l'utilisation d'une dose de charge d'Hulio chez les enfants âgés de moins de 6 ans (voir rubrique Propriétés pharmacocinétiques).
Il n'existe pas d'utilisation justifiée de l'adalimumab chez les enfants âgés de moins de 2 ans dans cette indication.
Une réévaluation annuelle des bénéfices et des risques associés au traitement continu à long terme est recommandée (voir rubrique Propriétés pharmacodynamiques).
D'autres dosages et/ou présentations d'Hulio peuvent être disponibles en fonction des besoins thérapeutiques de chaque patient.
Insuffisants rénaux et/ou hépatiques
L'adalimumab n'a pas été étudié dans ces populations de patients. Il n'est pas possible de recommander des posologies.
Mode d'administration
Hulio est administré en injection sous-cutanée. Les instructions complètes d'utilisation sont fournies dans la notice.
Un stylo de 40 mg et une seringue préremplie de 40 mg sont également disponibles pour permettre aux patients de s'injecter une dose complète de 40 mg.
Durée de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler. Conserver le flacon dans l'emballage extérieur à l'abri de la lumière.
2 ans
Précautions particulières de conservation :À conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler. Conserver le flacon dans l'emballage extérieur à l'abri de la lumière.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Aucune toxicité liée à la dose n'a été observée dans les essais
cliniques. La plus forte dose évaluée était constituée de doses
répétées de 10 mg/kg en IV, ce qui représente 15 fois environ la dose
recommandée.
Classe pharmacothérapeutique : Immunosuppresseurs, inhibiteurs du facteur de nécrose tumorale alpha (TNF-α). Code ATC : L04AB04
Hulio est un médicament biosimilaire. Des informations détaillées sont disponibles sur le site internet de l'Agence européenne des médicaments http://www.ema.europa.eu.
Mécanisme d'action
L'adalimumab se lie spécifiquement au TNF dont il neutralise la fonction biologique en bloquant son interaction avec les récepteurs du TNF p55 et p75 situés à la surface cellulaire.
L'adalimumab module aussi les réponses biologiques induites ou régulées par le TNF, y compris les variations des taux des molécules d'adhésion responsables de la migration des leucocytes (ELAM-1, VCAM-1 et ICAM-1 avec un IC50 de 0,1 - 0,2 nM).
Effets pharmacodynamiques
Après traitement par l'adalimumab chez des patients atteints de polyarthrite rhumatoïde, on a observé une diminution rapide du taux des marqueurs de la phase aiguë de l'inflammation (protéine réactive C [CRP], vitesse de sédimentation [VS]) et des cytokines sériques [IL-6] par rapport aux valeurs initialement observées. L'administration de l'adalimumab est également associée à une diminution des taux sériques des métalloprotéinases matricielles (MMP-1 et MMP-3) qui permettent le remodelage tissulaire responsable de la destruction cartilagineuse. Les patients traités par l'adalimumab présentent généralement une amélioration des signes hématologiques de l'inflammation chronique.
Une diminution rapide du taux de CRP a également été observée chez les patients atteints d'arthrite juvénile idiopathique polyarticulaire, de la maladie de Crohn, de rectocolite hémorragique et d'hidrosadénite suppurée après traitement par l'adalimumab. Chez les patients atteints de la maladie de Crohn, une réduction du nombre de cellules exprimant les marqueurs de l'inflammation dans le colon y compris une réduction significative de l'expression du TNFα a été observée. Des études endoscopiques sur la muqueuse intestinale ont mis en évidence une cicatrisation de la muqueuse chez les patients traités par l'adalimumab.
Efficacité et sécurité cliniques
Adultes atteints de polyarthrite rhumatoïde
L'adalimumab a fait l'objet d'études chez plus de 3 000 patients dans l'ensemble des essais cliniques dans la polyarthrite rhumatoïde (PR). L'efficacité et le profil de sécurité de l'adalimumab ont été évalués dans cinq études contrôlées randomisées, en double aveugle. Certains patients ont été traités pendant 120 mois.
L'étude I sur la PR a porté sur 271 patients atteints de polyarthrite rhumatoïde modérément à sévèrement active, âgés de 18 ans et plus et chez qui le traitement par au moins un traitement de fond (DMARD) avait échoué et chez qui le méthotrexate à la posologie de 12,5 à 25 mg/semaine (10 mg en cas d'intolérance au méthotrexate), s'était avéré insuffisamment efficace alors que la dose de méthotrexate était restée constante de 10 à 25 mg par semaine. Ces patients ont reçu 20, 40 ou 80 mg d'adalimumab ou un placebo toutes les deux semaines pendant 24 semaines.
L'étude II sur la PR a évalué 544 patients atteints de polyarthrite rhumatoïde modérément à sévèrement active, âgés de 18 ans et plus et chez qui le traitement par au moins un médicament anti- rhumatismal de fond avait échoué. Les patients ont été traités par 20 ou 40 mg d'adalimumab par voie sous-cutanée toutes les deux semaines en alternance avec un placebo, ou chaque semaine pendant 26 semaines ; un placebo a été administré toutes les semaines pendant la même durée. Aucun autre médicament anti-rhumatismal de fond n'était autorisé.
L'étude III sur la PR a évalué 619 patients atteints de polyarthrite rhumatoïde modérément à sévèrement active, âgés de 18 ans et plus qui n'avaient pas présenté de réponse au méthotrexate aux doses de 12,5 à 25 mg ou qui ne toléraient pas une dose de 10 mg de méthotrexate une fois par semaine. L'étude comportait trois groupes. Le premier a reçu des injections hebdomadaires de placebo pendant 52 semaines. Le deuxième a reçu 20 mg d'adalimumab toutes les semaines pendant 52 semaines Le troisième a été traité par 40 mg d'adalimumab toutes les deux semaines en alternance avec le placebo. Après la fin de la première période de 52 semaines, 457 patients ont été inclus dans une phase d'extension en ouvert au cours de laquelle l'adalimumab a été administré à la dose de 40 mg toutes les deux semaines en association au méthotrexate pendant 10 ans.
L'étude IV sur la PR a évalué principalement la tolérance chez 636 patients atteints de polyarthrite rhumatoïde d'activité modérée à sévère et âgés de 18 ans et plus. Les patients pouvaient n'avoir jamais reçu de médicament anti-rhumatismal de fond ou pouvaient poursuivre leur traitement rhumatologique préexistant pourvu que ce dernier soit stable depuis au moins 28 jours. Ces traitements comprenaient le méthotrexate, le léflunomide, l'hydroxychloroquine, la sulfasalazine et/ou les sels d'or. Les patientsaprès randomisation ont reçu soit 40 mg d'adalimumab, soit un placebo toutes les deux semaines pendant 24 semaines.
L'étude V sur la PR a évalué 799 patients adultes naïfs de méthotrexate ayant une polyarthrite rhumatoïde modérément à sévèrement active, récente (durée moyenne de la maladie inférieure à 9 mois). Cette étude a évalué l'efficacité de l'association de l'adalimumab à la dose de 40 mg toutes les deux semaines/méthotrexate, de l'adalimumab 40 mg toutes les deux semaines en monothérapie et une monothérapie de méthotrexate sur les signes et symptômes et le taux de progression des dommages structuraux dans la polyarthrite rhumatoïde, pendant 104 semaines. Après la fin de la première période de 104 semaines, 497 patients ont été inclus dans une phase d'extension en ouvert au cours de laquelle l'adalimumab a été administré à la dose de 40 mg toutes les deux semaines jusqu'à 10 ans.
Le critère de jugement principal des études I, II et III sur la PR, et le critère de jugement secondaire de l'étude IV sur la PR étaient le pourcentage de patients ayant obtenu une réponse ACR 20 à la semaine 24 ou 26. Le critère de jugement principal dans l'étude V sur la PR était le pourcentage de patients qui avaient obtenu une réponse ACR 50 à la semaine 52. Les études III et V sur la PR avaient un critère de jugement principal supplémentaire à 52 semaines à savoir le retard de progression de la maladie (attesté par les résultats radiologiques). L'étude III sur la PR avait aussi comme critère de jugement principal les modifications de la qualité de vie.
Réponse ACR
Le pourcentage de patients sous adalimumab qui ont obtenu une réponse ACR 20, 50 ou 70 a été cohérent dans les essais I, II et III sur la PR. Le tableau 8 résume les résultats obtenus à la posologie de 40 mg toutes les deux semaines.
Tableau 8 : Réponses ACR dans les essais contrôlés contre placebo (pourcentage de patients)
| Réponse | Étude Ia** sur la PR | Étude IIa** sur la PR | Étude IIIa** sur la PR | |||
| Placebo/MTXc n=60 | Adalimumabb/MTXc n=63 | Placebo n=110 | Adalimum abb n=113 | Placebo/MTXc n=200 | Adalimumab/MTXc n=207 | |
| ACR 20 | | | | | | |
| 6 mois | 13,3% | 65,1% | 19,1% | 46,0% | 29,5% | 63,3% |
| 12 mois | NA | NA | NA | NA | 24,0% | 58,9% |
| ACR 50 | | | | | | |
| 6 mois | 6,7% | 52,4% | 8,2% | 22,1% | 9,5% | 39,1% |
| 12 mois | NA | NA | NA | NA | 9,5% | 41,5% |
| ACR 70 | | | | | | |
| 6 mois | 3,3% | 23,8% | 1,8% | 12,4% | 2,5% | 20,8% |
| 12 mois | NA | NA | NA | NA | 4,5% | 23,2% |
b 40 mg d'adalimumab administré toutes les deux semaines
c MTX = méthotrexate
** p < 0,01, adalimumab versus placebo
Dans les études I-IV sur la PR, les composantes individuelles des critères de réponse de l'ACR (nombre d'articulations sensibles et tuméfiées, évaluation par le médecin et le patient de l'activité de la maladie et de la douleur, indice d'invalidité [HAQ] et valeurs de la CRP [mg/dl]) ont été améliorées à 24 ou 26 semaines par rapport au placebo. Dans l'étude III sur la PR, ces améliorations se sont maintenues tout au long des 52 semaines.
Dans la phase d'extension en ouvert de l'étude III sur la PR, les taux de réponse ACR ont été maintenus chez la plupart des patients suivis jusqu'à 10 ans. Sur 207 patients qui avaient été randomisés dans le bras adalimumab 40 mg toutes les 2 semaines, 114 patients ont poursuivi l'adalimumab à la dose de 40 mg toutes les deux semaines pendant 5 ans. Parmi ces patients, 86 (75,4%) ont eu une réponse ACR 20 ; 72 (63,2%) ont eu une réponse ACR 50 et 41 (36%) ont eu une réponse ACR 70. Sur 207 patients, 81 ont poursuivi l'adalimumab à la dose de 40 mg toutes les deux semaines pendant 10 ans. Parmi ces patients, 64 (79,0%) ont eu une réponse ACR 20 ;56 (69,1%) ont eu une réponse ACR 50 et 43 (53,1%) ont eu une réponse ACR 70.
Dans l'étude IV sur la PR, la réponse ACR 20 des patients traités par l'adalimumab en plus des soins habituels a été significativement meilleure que chez les patients recevant le placebo plus les soins habituels (p < 0,001).
Dans les études I - IV sur la PR, les réponses ACR 20 et 50 des patients traités par l'adalimumab ont été statistiquement significatives par rapport au placebo dès la première ou la deuxième semaine de traitement.
Dans l'étude V sur la PR chez des patients ayant une polyarthrite rhumatoïde récente, naïfs de méthotrexate, un traitement associant adalimumab et méthotrexate a conduit à des réponses ACR plus rapides et significativement plus importantes qu'avec le méthotrexate seul et l'adalimumab seul, à la semaine 52 et les réponses étaient maintenues à la semaine 104 (voir tableau 9).
Tableau 9 : Réponses ACR dans l'étude V sur la PR (pourcentage de patients)
| Réponse | MTX n=257 | Adalimumab n=274 | Adalimumab/MTX n=268 | Valeur de pa | Valeur de pb | Valeur de pc |
| ACR 20 | | | | | | |
| Semaine 52 | 62,6% | 54,4% | 72,8% | 0,013 | < 0,001 | 0,043 |
| Semaine 104 | 56,0% | 49,3% | 69,4% | 0,002 | < 0,001 | 0,140 |
| ACR 50 | | | | | | |
| Semaine 52 | 45,9% | 41,2% | 61,6% | < 0,001 | < 0,001 | 0,317 |
| Semaine 104 | 42,8% | 36,9% | 59,0% | < 0,001 | < 0,001 | 0,162 |
| ACR 70 | | | | | | |
| Semaine 52 | 27,2% | 25,9% | 45,5% | < 0,001 | < 0,001 | 0,656 |
| Semaine 104 | 28,4% | 28,1% | 46,6% | < 0,001 | < 0,001 | 0,864 |
a La valeur de p résulte de la comparaison appariée des traitements par adalimumab seul et par l'association adalimumab/méthotrexate par le test U de Mann-Whitney.
c La valeur de p résulte de la comparaison appariée des traitements par adalimumab seul et par méthotrexate seul par le test U de Mann-Whitney.
Dans la phase d'extension en ouvert de l'étude V sur la PR, les taux de réponse ACR ont été maintenus chez les patients suivis jusqu'à 10 ans. Sur 542 patients qui avaient été randomisés dans le bras adalimumab 40 mg toutes les 2 semaines, 170 patients ont poursuivi l'adalimumab à la dose de 40 mg toutes les deux semaines pendant 10 ans. Parmi ces patients, 154 (90,6%) ont eu uneréponse ACR 20 ; 127 (74,7%) ont eu une réponse ACR 50 et 102 (60,0%) ont eu une réponse ACR 70.
À la semaine 52, 42,9% des patients qui avaient reçu l'association adalimumab/méthotrexate étaient en rémission clinique (DAS 28 (CRP) < 2,6) comparativement à 20,6% des patients ayant reçu le méthotrexate seul et 23,4% des patients ayant reçu l'adalimumab seul. Le traitement par l'association adalimumab/méthotrexate était cliniquement et statistiquement supérieur au méthotrexate (p < 0,001) et à l'adalimumab en monothérapie (p < 0,001) dans l'obtention d'un état d'activité basse de la maladie pour les patients chez qui une polyarthrite rhumatoïde modérée à sévère avait été récemment diagnostiquée. La réponse pour les deux bras de monothérapie était similaire (p = 0,447). Sur 342 patients initialement randomisés pour recevoir l'adalimumab seul ou l'association adalimumab/méthotrexate qui ont été inclus dans l'étude d'extension en ouvert, 171 patients ont terminé 10 ans de traitement par l'adalimumab. Parmi ces patients, 109 patients (63,7%) étaient en rémission à 10 ans.
Réponse radiographique
Dans l'étude III sur la PR, dans laquelle les patients traités par l'adalimumab avaient une polyarthrite rhumatoïde d'une durée moyenne de 11 ans environ, les dommages structuraux articulaires ont été évalués par radiographie et exprimés en matière de modification du score total de Sharp (STS) et de ses composants, le score d'érosion et le score de pincement articulaire. Les patients traités par l'adalimumab associé au méthotrexate ont présenté une progression significativement moindre que les patients recevant seulement du méthotrexate à 6 et 12 mois (voir tableau 10).
Dans l'extension en ouvert de l'étude III dans la PR, le ralentissement de la progression des dommages structuraux est maintenu à 8 et 10 ans pour une partie des patients. À 8 ans, 81 des 207 patients traités dès le début par 40 mg d'adalimumab une semaine sur deux ont été évalués par radiographie. Parmi ces patients, 48 patients n'ont pas présenté de progression des dommages structuraux définie par une modification du score total de Sharp modifié de 0,5 ou moins par rapport à la valeur de base. À 10 ans, 79 des 207 patients traités dès le début par 40 mg d'adalimumab une semaine sur deux ont été évalués par radiographie. Parmi ces patients, 40 patients n'ont pas présenté de progression des dommages structuraux définie par une modification du score total de Sharp modifié de 0,5 ou moins par rapport à la valeur de base.
Tableau 10 : Valeurs moyennes des modifications radiographiques sur 12 mois dans l'étude III sur la PR
| | Placebo/MTXa | Adalimumab/MTX 40 mg toutes les deux semaines | Placebo/MTX- adalimumab/MTX (intervalle de confiance à 95%b) | Valeur p |
| Score total de Sharp | 2,7 | 0,1 | 2,6 (1,4 ; 3,8) | < 0,001c |
| Score d'érosion | 1,6 | 0,0 | 1,6 (0,9 ; 2,2) | < 0,001 |
| Score de pincement articulaire (JSN)d | 1,0 | 0,1 | 0,9 (0,3 ; 1,4) | 0,002 |
b Intervalles de confiance à 95% des différences de variations des scores entre méthotrexate et adalimumab.
c D'après les analyses de rang
d JSN, Joint Space Narrowing
Dans l'étude V sur la PR, les dommages structuraux articulaires ont été évalués par radiographie et exprimés en termes de variation du score total de Sharp (voir tableau 11).
Tableau 11 : Valeurs moyennes des modifications radiographiques à la semaine 52 dans l'étude V sur la PR
| | MTX n=257 (Intervalle de confiance à 95%) | Adalimumab n=274 (Intervalle de confiance à 95%) | Adalimumab/MTX n=268 (Intervalle de confiance à 95%) | Valeur de pa | Valeur de pb | Valeur de pc |
| Score total de Sharp | 5,7 (4,2-7,3) | 3,0 (1,7-4,3) | 1,3 (0,5-2,1) | < 0,001 | 0,0020 | < 0,001 |
| Score d'érosion | 3,7 (2,7-4,7) | 1,7 (1,0-2,4) | 0,8 (0,4-1,2) | < 0,001 | 0,0082 | < 0,001 |
| Score de pincement articulaire (JSN) | 2,0 (1,2-2,8) | 1,3 (0,5-2,1) | 0,5 (0-1,0) | < 0,001 | 0,0037 | 0,151 |
b La valeur de p résulte de la comparaison appariée des traitements par adalimumab seul et par l'association adalimumab/méthotrexate par le test U de Mann-Whitney.
c La valeur de p résulte de la comparaison appariée des traitements par adalimumab seul et par méthotrexate seul par le test U de Mann-Whitney.
À la suite de 52 et 104 semaines de traitement, le pourcentage de patients sans progression (variation du score total de Sharp modifié par rapport à la valeur de base ≤ 0,5) était significativement supérieur avec le traitement par l'association adalimumab/méthotrexate (respectivement 63,8% et 61,2%) comparativement au méthotrexate en monothérapie (respectivement 37,4% et 33,5%, p < 0,001) et l'adalimumab en monothérapie (respectivement 50,7%, p < 0,002 et 44,5%, p < 0,001).
Dans la phase d'extension en ouvert de l'étude V sur la PR, la variation moyenne du score total de Sharp modifié à 10 ans par rapport à la valeur de base a été de 10,8 chez les patients randomisés initialement pour recevoir le méthotrexate en monothérapie, 9,2 chez les patients randomisés initialement pour recevoir l'adalimumab en monothérapie et 3,9 chez les patients randomisés initialement pour recevoir l'association adalimumab/méthotrexate. Les proportions correspondantes de patients ne présentant pas de progression radiographique ont été respectivement de 31,3%, 23,7% et 36,7%.
Qualité de vie et capacités fonctionnelles
La qualité de vie en rapport avec la santé et la fonction physique ont été évaluées au moyen de l'indice d'invalidité du Questionnaire d'Évaluation de l'état de Santé (Health Assessment Questionnaire, HAQ) dans les quatre essais originels adéquats et correctement contrôlés, et constituait un critère principal de jugement pré-spécifié à la semaine 52 dans l'étude III sur la PR. Comparativement au placebo, toutes les doses/schémas posologiques d'administration d'adalimumab ont entraîné une amélioration statistiquement significative plus importante de l'indice d'invalidité du HAQ entre l'examen initial et le 6e mois dans les quatre études et il en a été de même à la semaine 52 dans l'étude III sur la PR. Dans les quatre études, les résultats des scores de la Short Form Health Survey (SF-36) confirment ces observations pour toutes les doses/schémas posologiques d'administration d'adalimumab, avec des valeurs des composantes physiques (PCS) statistiquement significatives, ainsi que des scores de douleur et de vitalité statistiquement significatifs pour la dose de 40 mg toutes les deux semaines. Dans les trois études dans lesquelles elle a été prise en compte (études I, III et IV sur la PR), on a observé une diminution statistiquement significative de la fatigue mesurée à l'aide des scores d'évaluation fonctionnelle de traitement pour maladie chronique (FACIT).
Dans l'étude III sur la PR, la plupart des patients ayant obtenu une amélioration des capacités fonctionnelles et ayant poursuivi le traitement ont maintenu cette amélioration jusqu'à la semaine 520 (120 mois) du traitement en ouvert. L'amélioration de la qualité de vie a été mesurée jusqu'à la semaine 156 (36 mois) et l'amélioration a été maintenue au cours de cette période.
Dans l'étude V sur la PR, l'amélioration de l'indice d'invalidité HAQ et la composante physique du SF-36 s'est montrée beaucoup plus importante (p < 0,001) pour l'association adalimumab/méthotrexate par rapport à la monothérapie de méthotrexate et la monothérapie d'adalimumab à la semaine 52, et s'est maintenue jusqu'à la semaine 104. Parmi les 250 patients ayant terminé l'étude d'extension en ouvert, l'amélioration des capacités fonctionnelles s'est maintenue au cours des 10 ans de traitement.
Psoriasis en plaques chez l'adulte
L'efficacité et la tolérance de l'adalimumab ont été évaluées lors d'études randomisées menées en double aveugle chez des patients adultes atteints de psoriasis chronique en plaques (intéressant ≥ 10% de la surface corporelle, avec un indice de sévérité PASI ≥ 12 ou ≥ 10) qui étaient candidats à un traitement systémique ou une photothérapie. Au total, 73% des patients recrutés dans les études I et II sur le psoriasis avaient déjà reçu un traitement systémique ou une photothérapie. L'efficacité et la tolérance de l'adalimumab ont également été étudiées chez des patients adultes atteints de psoriasis chronique en plaques modéré à sévère avec une atteinte concomitante des mains et/ou des pieds qui étaient candidats à un traitement systémique dans une étude randomisée en double aveugle (étude III sur le psoriasis).
L'étude I sur le psoriasis (REVEAL) a porté sur 1 212 patients pendant trois périodes de traitement. Durant la période A, les patients recevaient un placebo ou l'adalimumab à la dose initiale de 80 mg, suivie de 40 mg une semaine sur deux à partir d'une semaine après la dose initiale. Au bout de 16 semaines de traitement, les patients ayant obtenu au minimum une réponse PASI 75 (amélioration d'au moins 75% du score PASI par rapport aux valeurs initiales), entraient dans la période B et recevaient 40 mg d'adalimumab en ouvert une semaine sur deux. Les patients dont la réponse restait ≥ PASI 75 à la semaine 33 et qui avaient été initialement randomisés pour recevoir le traitement actif pendant la période A, ont à nouveau été randomisés pendant la période C pour recevoir 40 mg d'adalimumab une semaine sur deux ou un placebo pendant 19 semaines supplémentaires. Dans tous les groupes de traitement, le score PASI initial moyen était de 18,9 et le score PGA était compris entre« modéré » (53% des sujets inclus) et « sévère » (41%), voire « très sévère » (6%).
L'étude II sur le psoriasis (CHAMPION) a comparé l'efficacité et la tolérance d'adalimumab à celles du méthotrexate et d'un placebo chez 271 patients. Les patients ont reçu un placebo, une dose initiale de MTX à 7,5 mg, augmentée ensuite jusqu'à la semaine 12, la dose maximale étant de 25 mg, ou bien une dose initiale de 80 mg d'adalimumab suivie de 40 mg une semaine sur deux (en commençant une semaine après la semaine initiale) pendant 16 semaines. On ne dispose d'aucune donnée concernant la comparaison entre l'adalimumab et le MTX au-delà de 16 semaines de traitement. Chez les patients sous MTX ayant atteint une réponse ≥ PASI 50 à la semaine 8 et/ou 12, la posologie n'était pas augmentée davantage. Dans tous les groupes de traitement, le score PASI initial moyen était de 19,7 et le score PGA initial allait de « léger » (< 1%) à « modéré » (48%), « sévère » (46%) et « très sévère » (6%).
Les patients ayant participé aux études de phase II et III dans le psoriasis étaient éligibles pour entrer dans une étude d'extension en ouvert, dans laquelle l'adalimumab était administré pendant au moins 108 semaines supplémentaires.
Un des principaux critères d'évaluation des études I et II sur le psoriasis était le pourcentage de patients ayant atteint une réponse PASI 75 entre l'inclusion et la semaine 16 (voir Tableaux 12 et 13).
Tableau 12 : Étude I sur le psoriasis (REVEAL) - Résultats d'efficacité à 16 semaines
| | Placebo N = 398 n (%) | Adalimumab 40 mg toutes les deux semaines N = 814 n (%) |
| ≥ PASI 75a | 26 (6,5) | 578 (70,9)b |
| PASI 100 | 3 (0,8) | 163 (20,0)b |
| PGA : clair/minimal | 17 (4,3) | 506 (62,2)b |
b p < 0,001, adalimumab versus placebo
Tableau 13 : Étude II sur le psoriasis (CHAMPION) - Résultats d'efficacité à 16 semaines
| | Placebo N = 53 n (%) | MTX N = 110 n (%) | Adalimumab 40 mg toutes les deux semaines N = 108 n (%) |
| ≥ PASI 75 | 10 (18.9) | 39 (35.5) | 86 (79,6)a,b |
| PASI 100 | 1 (1.9) | 8 (7.3) | 18 (16,7)c,d |
| PGA : clair/minimal | 6 (11.3) | 33 (30.0) | 79 (73,1)a,b |
b p < 0,001, adalimumab versus méthotrexate
c p < 0,01, adalimumab versus placebo
d p < 0,05, adalimumab versus méthotrexate
Dans l'étude I sur le psoriasis, 28% des patients ayant présenté une réponse PASI 75 et randomisés à nouveau pour recevoir le placebo à la semaine 33, et 5% de ceux poursuivant le traitement par l'adalimumab (p < 0,001), ont présenté « une diminution de la réponse appropriée » (score PASI entre la semaine 33 et la semaine 52 (incluse) se traduisant par une réponse < PASI 50 par rapport à l'inclusion, avec un minimum d'augmentation de 6 points du score PASI par rapport à la semaine 33). Parmi les patients présentant une diminution de la réponse appropriée après la nouvelle randomisation dans le groupe placebo et ensuite recrutés dans l'étude d'extension en ouvert, 38% (25/66) et 55% (36/66) ont retrouvé une réponse PASI 75 au bout de respectivement 12 et 24 semaines.
Un total de 233 patients répondeurs PASI 75 à la semaine 16 et à la semaine 33 ont reçu un traitement en continu par l'adalimumab pendant 52 semaines dans l'étude I et ont poursuivi le traitement par l'adalimumab dans l'étude d'extension en ouvert. Le taux de réponse PASI 75 et PGA clair ou minimal chez ces patients étaient respectivement de 74,7% et 59,0%, après 108 semaines supplémentaires de traitement en ouvert (total de 160 semaines). Dans une analyse où tous les patients sortis d'essai pour effets indésirables ou pour manque d'efficacité, ou pour lesquels la dose a été augmentée, ont été considérés comme non-répondeurs, le taux de réponse PASI 75 et PGA clair ou minimal chez ces patients étaient respectivement de 69,6% et 55,7%, après 108 semaines supplémentaires de traitement en ouvert (total de 160 semaines).
Un total de 347 patients répondeurs stables ont participé à une évaluation d'interruption de traitement et de retraitement dans une étude d'extension en ouvert. Durant la période d'interruption de traitement, les symptômes du psoriasis sont réapparus au cours du temps avec un délai médian de rechute (régression vers un PGA « modéré » ou plus sévère) d'environ 5 mois. Aucun patient n'a présenté de rebond durant la phase d'interruption de traitement. 76,5% (218/285) des patients qui sont entrés dans la période de retraitement ont eu une réponse PGA « clair » ou « minimal » après 16 semaines de retraitement, indépendamment du fait qu'ils aient rechuté ou non durant l'interruption de traitement (69,1% [123/178] pour les patients qui ont rechuté durant la période d'interruption et 88,8% [95/107] pour les patients qui n'ont pas rechuté durant la période d'interruption). Un profil de tolérance similaire a été observé durant le retraitement et avant l'interruption de traitement.
L'index dermatologique de qualité de vie DLQI (Dermatology Life Quality Index) a mis en évidence des améliorations significatives à la semaine 16 par rapport à l'inclusion, comparativement au placebo (études I et II) et au MTX (étude II). Dans l'étude I, les améliorations des scores résumés des composantes physiques et psychologiques du SF-36 étaient également significatives par rapport au placebo.
Dans une étude d'extension en ouvert chez les patients ayant dû augmenter les doses (de 40 mg une semaine sur deux à 40 mg toutes les semaines) en raison d'une réponse PASI inférieure à 50%, 26,4% (92/349) et 37,8% (132/349) des patients ont atteint une réponse PASI 75 à la semaine 12 et à la semaine 24, respectivement.
L'étude III dans le psoriasis (REACH) a comparé l'efficacité et la tolérance d'adalimumab par rapport au placebo chez 72 patients présentant un psoriasis chronique en plaques modéré à sévère avec une atteinte concomitante des mains et/ou des pieds. Les patients ont reçu une dose initiale de 80 mg d'adalimumab suivi par 40 mg toutes les 2 semaines (en commençant une semaine après la dose initiale) ou le placebo pendant 16 semaines. À la semaine 16, une proportion statistiquement significative plus importante de patients ayant reçu l'adalimumab ont atteint un PGA « clair » ou « pratiquement clair » pour les mains et/ou les pieds par rapport à ceux qui ont reçu le placebo (30,6% versus 4,3%, respectivement [p = 0,014]).
L'étude IV dans le psoriasis a comparé l'efficacité et la tolérance de l'adalimumab par rapport au placebo chez 217 patients adultes atteints de psoriasis unguéal modéré à sévère. Les patients ont reçu une dose initiale de 80 mg d'adalimumab, suivie par 40 mg toutes les deux semaines (en commençant une semaine après la dose initiale) ou un placebo pendant 26 semaines suivi d'un traitement par l'adalimumab en ouvert pendant 26 semaines supplémentaires. L'évaluation du psoriasis unguéal a été faite sur la base de l'indice modifié de sévérité du psoriasis unguéal (mNAPSI, Modified Nail Psoriasis Severity Index), de l'évaluation globale par le médecin de la sévérité du psoriasis des ongles des mains (PGA-F, Physician's Global Assessment of Fingernail Psoriasis) et de l'indice de sévérité du psoriasis unguéal (NAPSI, Nail Psoriasis Severity Index) (voir Tableau 14). L'adalimumab a démontré un bénéfice dans le traitement des patients atteints de psoriasis unguéal présentant différents degrés d'atteinte cutanée (SCA ≥ 10% [60% des patients] et SCA < 10% et ≥ 5% [40% des patients]).
Tableau 14 : Étude IV sur le psoriasis - Résultats d'efficacité à 16, 26 et 52 semaines
| Critère de jugement | Semaine 16 Phase contrôlée versus placebo | Semaine 26 Phase contrôlée versus placebo | Semaine 52 Phase en ouvert | ||
| Placebo N = 108 | Adalimumab 40 mg toutes les deux semaines N = 109 | Placebo N = 108 | Adalimumab 40 mg toutes les deux semaines N = 109 | Adalimumab 40 mg toutes les deux semaines N = 80 | |
| ≥ mNAPSI 75 (%) | 2,9 | 26,0a | 3,4 | 46,6a | 65,0 |
| PGA-F clair/minimal et ≥ 2 grades d'amélioration (%) | 2,9 | 29,7a | 6,9 | 48,9a | 61,3 |
| Changement, en pourcentage, de l'indice total de sévérité du psoriasis unguéal (NAPSI) (%) | -7,8 | -44,2a | -11,5 | -56,2a | -72,2 |
Les patients traités par l'adalimumab ont montré des améliorations statistiquement significatives du DLQI à la semaine 26 par rapport au groupe placebo.
Hidrosadénite suppurée de l'adulte
La sécurité et l'efficacité d'adalimumab ont été évaluées au cours d'études randomisées, en double aveugle, contrôlées par rapport au placebo et d'une étude d'extension en ouvert chez des patients adultes atteints d'hidrosadénite suppurée (HS) modérée à sévère ayant présenté une intolérance, une contre-indication ou une réponse insuffisante à un traitement antibiotique systémique de 3 mois minimum. Les patients des études HS-I et HS-II présentaient une maladie de stade II ou III selon la classification de Hurley, avec au moins 3 abcès ou nodules inflammatoires.
L'étude
HS-I (PIONEER I) a évalué 307 patients sur 2 périodes de traitement. Au
cours de la période A, les patients recevaient le placebo ou
l'adalimumab à une dose initiale de 160 mg à la semaine 0, puis 80 mg à
la semaine 2, et 40 mg toutes les semaines de la semaine 4 à la semaine
11.
L'utilisation
concomitante d'antibiotiques n'était pas autorisée au cours de l'étude.
Après 12 semaines de traitement, les patients traités par l'adalimumab
au cours de la période A ont été de nouveau randomisés dans la période
B dans l'un des 3 groupes de traitement (adalimumab 40 mg toutes les
semaines, adalimumab 40 mg toutes les deux semaines, ou placebo de la
semaine 12 à la semaine 35). Les patients randomisés pour recevoir le
placebo au cours de la période A ont été affectés au groupe de
l'adalimumab 40 mg toutes les semaines de la période B.
L'étude HS-II (PIONEER II) a évalué 326 patients sur 2 périodes de traitement. Au cours de la période A, les patients recevaient le placebo ou l'adalimumab à une dose initiale de 160 mg à la semaine 0, puis 80 mg à la semaine 2, et 40 mg toutes les semaines de la semaine 4 à la semaine 11. Au cours de l'étude, 19,3% des patients ont poursuivi le traitement antibiotique oral qu'ils prenaient à l'inclusion. Après 12 semaines de traitement, les patients traités par l'adalimumab au cours de la période A ont été de nouveau randomisés dans la période B dans l'un des 3 groupes de traitement (adalimumab 40 mg toutes les semaines, adalimumab 40 mg toutes les deux semaines, ou placebo de la semaine 12 à la semaine 35). Les patients randomisés pour recevoir le placebo au cours de la période A ont été affectés au groupe placebo de la période B.
Les patients des études HS-I et HS-II étaient éligibles à l'inclusion dans une étude d'extension en ouvert au cours de laquelle l'adalimumab 40 mg était administré toutes les semaines. L'exposition moyenne dans toute la population traitée par l'adalimumab a été de 762 jours. Tout au long des 3 études, les patients ont utilisé quotidiennement un antiseptique local sur leurs lésions.
Réponse clinique
La réduction des lésions inflammatoires et la prévention de l'aggravation des abcès et des fistules drainantes ont été évaluées à l'aide du score de réponse clinique dans l'hidrosadénite suppurée (HiSCR : réduction d'au-moins 50% du nombre total d'abcès et de nodules inflammatoires sans augmentation du nombre d'abcès ni du nombre de fistules drainantes par rapport aux valeurs à l'inclusion). La réduction de la douleur cutanée liée à l'HS a été évaluée à l'aide d'une échelle numérique chez les patients de l'étude qui présentaient à l'inclusion un score initial supérieur ou égal à 3 sur une échelle de 11 points.
À la semaine 12, il y a eu significativement plus de répondeurs HiSCR dans le groupe de l'adalimumab comparativement au groupe placebo. À la semaine 12 de l'étude HS-II, un pourcentage significativement plus élevé de patients a obtenu une réduction cliniquement significative de la douleur liée à l'HS (voir tableau 15). Les patients traités par l'adalimumab présentaient une réduction significative du risque de poussée de la maladie au cours des 12 premières semaines de traitement.
Tableau 15 : Résultats d'efficacité à 12 semaines, études HS-I et HS-II
| | Étude HS-I | Étude HS-II | ||
| Placebo | Adalimumab 40 mg par semaine | Placebo | Adalimumab 40 mg par semaine | |
| Réponse clinique dans l'hidrosadénite suppurée (HiSCR)a | N = 154 40 (26,0%) | N = 153 64 (41,8%)* | N = 163 45 (27,6%) | N = 163 96 (58,9%)*** |
| Réduction ≥ 30% de la douleur cutanéeb | N = 109 27 (24,8%) | N = 122 34 (27,9%) | N = 111 23 (20,7%) | N = 105 48 (45,7%)*** |
a Chez tous les patients randomisés.
b Chez les patients présentant un score de la douleur cutanée liée à l'HS à l'inclusion ≥ 3, sur la base d'une échelle numérique de 0 à 10, avec 0 = aucune douleur cutanée, 10 = pire douleur cutanée imaginable.
Le traitement par l'adalimumab 40 mg toutes les semaines a réduit significativement le risque d'aggravation des abcès et fistules drainantes. Au cours des 12 premières semaines des études HS-I et HS-II, environ deux fois plus de patients du groupe placebo ont présenté une aggravation des abcès (23,0% contre 11,4%, respectivement) et des fistules drainantes (30,0% contre 13,9%, respectivement), comparativement aux groupes de l'adalimumab.
Par rapport au placebo, des améliorations plus importantes entre l'inclusion et la semaine 12 ont été observées en matière de qualité de vie liée à la santé propre à la peau mesurée par l'indice de qualité de vie dermatologique (DLQI ; études HS-I et HS-II), de satisfaction globale du patient par rapport à son traitement médicamenteux mesurée par le questionnaire de satisfaction relative au traitement médicamenteux (TSQM ; études HS-I et HS-II), et de santé physique mesurée par la composante physique du score SF-36 (étude HS-I).
Chez les patients présentant au moins une réponse partielle à la semaine 12, traités par l'adalimumab 40 mg administré toutes les semaines, le taux de réponse clinique HiSCR à la semaine 36 était plus élevé chez les patients recevant l'adalimumab toutes les semaines par rapport aux patients chez lesquels la fréquence d'administration était réduite à toutes les deux semaines ou chez lesquels le traitement était interrompu (voir tableau 16).
Tableau 16 : Pourcentage de patientsa répondeurs HiSCRb aux semaines 24 et 36 après re- randomisation des traitements à la semaine 12, patients issus du groupe de l'adalimumab toutes les semaines
| | Placebo (arrêt du traitement) N = 73 | Adalimumab 40 mg toutes les deux semaines N = 70 | Adalimumab 40 mg par semaine N = 70 |
| Semaine 24 | 24 (32,9%) | 36 (51,4%) | 40 (57,1%) |
| Semaine 36 | 22 (30,1%) | 28 (40,0%) | 39 (55,7%) |
b Les patients répondant aux critères spécifiés dans le protocole en matière de perte de réponse ou d'absence d'amélioration devaient sortir des études et étaient comptabilisés comme non- répondeurs.
Parmi les patients présentant au moins une réponse partielle à la semaine 12 et traités en continu par l'adalimumab toutes les semaines, le taux de répondeurs HiSCR à la semaine 48 était de 68,3% et de 65,1% à la semaine 96. Un traitement à plus long terme par l'adalimumab 40 mg une fois par semaine pendant 96 semaines n'a identifié aucun nouveau signal de sécurité.
Parmi les patients dont le traitement par l'adalimumab a été interrompu à la semaine 12 dans les études HS-I et HS-II, le taux de répondeurs HiSCR 12 semaines après la ré-introduction d'adalimumab 40 mg toutes les semaines était revenu à des valeurs semblables à celles observées avant l'interruption du traitement (56,0%).
Maladie de Crohn de l'adulte
La tolérance et l'efficacité d'adalimumab ont été évaluées chez plus de 1 500 patients ayant une maladie de Crohn active modérée à sévère (indice d'activité de la maladie de Crohn (CDAI [Crohn's Disease Activity Index]) ≥ 220 et ≤ 450) dans des études randomisées, en double aveugle, contrôlées par rapport au placebo. Des doses stables concomitantes d'aminosalicylés, de corticoïdes et/ou d'immunomodulateurs étaient autorisées et 80% des patients ont continué à recevoir au moins un de ces médicaments.
L'induction d'une rémission clinique (définie par un indice CDAI < 150) a été évaluée dans deux études, l'étude I sur la MC (CLASSIC I) et l'étude II sur la MC (GAIN). Dans l'étude I sur la MC, 299 patients non précédemment traités par un anti-TNF ont été randomisés dans l'un des quatre groupes de traitement de l'étude ; placebo aux semaines 0 et 2, 160 mg d'adalimumab à la semaine 0 et 80 mg à la semaine 2, 80 mg à la semaine 0 et 40 mg à la semaine 2, et 40 mg à la semaine 0 et 20 mg à la semaine 2. Dans l'étude II sur la MC, 325 patients ne répondant plus ou étant intolérants à l'infliximab ont été randomisés pour recevoir soit 160 mg d'adalimumab à la semaine 0 et 80 mg à la semaine 2, soit un placebo aux semaines 0 et 2. Les non-répondeurs primaires ont été exclus des études et ces patients n'ont par conséquent pas fait l'objet d'autres évaluations.
Le maintien de la rémission clinique a été évalué dans l'étude III sur la MC (CHARM). Dans l'étude III sur la MC, 854 patients ont reçu en ouvert 80 mg à la semaine 0 et 40 mg à la semaine 2. À la semaine 4, les patients ont été randomisés pour recevoir soit 40 mg toutes les deux semaines ou 40 mg toutes les semaines, soit un placebo pour une durée totale de 56 semaines. Les patients présentant une réponse clinique (diminution de l'indice CDAI ≥ 70) à la semaine 4 ont été stratifiés et analysés séparément de ceux n'ayant pas présenté de réponse clinique à la semaine 4. La diminution progressive des corticoïdes était autorisée après la semaine 8.
Les taux d'induction d'une rémission et de réponse enregistrés dans les études I et II sur la MC sont présentés dans le tableau 17.
Tableau 17 : Induction d'une rémission clinique et d'une réponse clinique (pourcentage de patients)
| | Étude I sur la MC : patients naïfs d'infliximab | Étude II sur la MC : patients précédemment traités par l'infliximab | |||
| | Placebo N = 74 | Adalimumab 80/40 mg N = 75 | Adalimumab 160/80 mg N = 76 | Placebo N = 166 | Adalimumab 160/80 mg N = 159 |
| Semaine 4 | | | | | |
| Rémission clinique | 12% | 24% | 36%* | 7% | 21%* |
| Réponse clinique (CR-100) | 24% | 37% | 49%** | 25% | 38%** |
* p < 0,001
** p < 0,01
Des taux de rémission similaires ont été observés pour les schémas d'induction 160/80 mg et 80/40 mg à la semaine 8, et les événements indésirables ont été plus fréquents dans le groupe 160/80 mg.
Dans l'étude III sur la MC, 58% (499/854) des patients présentaient une réponse clinique à la semaine 4 et ont été évalués dans l'analyse principale. Parmi les patients présentant une réponse clinique à la semaine 4, 48% avaient été préalablement exposés à un autre traitement anti-TNF. Les taux de maintien de la rémission et de réponse sont présentés dans le tableau 18. Les résultats de rémission clinique sont restés relativement constants, indépendamment de l'exposition antérieure éventuelle à un anti-TNF.
Les hospitalisations et les interventions chirurgicales liées à la maladie ont été réduites de manière statistiquement significative avec l'adalimumab comparé au placebo à la semaine 56.
Tableau 18 : Maintien de la rémission clinique et de la réponse clinique (pourcentage de patients)
| | Placebo | 40 mg d'adalimumab toutes les deux semaines | 40 mg d'adalimumab toutes les semaines |
| Semaine 26 | N = 170 | N = 172 | N = 157 |
| Rémission clinique | 17% | 40%* | 47%* |
| Réponse clinique (CR-100) | 27% | 52%* | 52%* |
| Patients en rémission sans corticoïdes depuis ≥ 90 joursa | 3% (2/66) | 19% (11/58)** | 15% (11/74)** |
| Semaine 56 | N = 170 | N = 172 | N = 157 |
| Rémission clinique | 12% | 36%* | 41%* |
| Réponse clinique (CR-100) | 17% | 41%* | 48%* |
| Patients en rémission sans corticoïdes depuis ≥ 90 joursa | 5% (3/66) | 29% (17/58)* | 20% (15/74)** |
* p < 0,02 pour l'adalimumab par rapport au placebo, comparaisons appariées des pourcentages
a Parmi ceux initialement traités par corticoïdes
Parmi les patients non répondeurs à la semaine 4, 43% des patients recevant un traitement d'entretien par l'adalimumab ont répondu à la semaine 12 contre 30% des patients recevant le placebo en traitement d'entretien. Ces résultats suggèrent que certains patients n'ayant pas répondu à la semaine 4 bénéficient de la poursuite du traitement d'entretien jusqu'à la semaine 12. La poursuite du traitement au-delà de 12 semaines n'est pas significativement associée à plus de réponses (voir rubrique Posologie et mode d'administration).
117/276 patients de l'étude I sur la MC et 272/777 patients des études II et III sur la MC ont été suivis pendant au moins 3 ans de traitement en ouvert par l'adalimumab. 88 et 189 patients, respectivement, sont restés en rémission clinique. La réponse clinique (CR-100) a été maintenue chez 102 et233 patients, respectivement.
Qualité de vie
Dans les études I et II sur la MC, une amélioration statistiquement significative du score total du questionnaire sur les maladies inflammatoires de l'intestin (IBDQ) spécifique de la maladie a été obtenue à la semaine 4 chez les patients traités par l'adalimumab 80/40 mg et 160/80 mg par rapport au placebo, et également aux semaines 26 et 56 dans l'étude III sur la MC ainsi que dans tous les groupes traités par l'adalimumab par rapport au placebo.
Uvéite chez l'adulte
La tolérance et l'efficacité de l'adalimumab ont été évaluées chez des patients adultes atteints d'uvéite non infectieuse, intermédiaire, postérieure et de panuvéite, à l'exclusion des patients présentant une uvéite antérieure isolée, dans deux études randomisées, en double aveugle, contrôlées contre placebo (UV I et II). Les patients recevaient un placebo ou l'adalimumab à la dose initiale de 80 mg puis de 40 mg toutes les deux semaines en commençant une semaine après l'administration de la première dose. L'administration concomitante d'un immunosuppresseur non biologique à dose stable était autorisée.
L'étude UV I a évalué 217 patients présentant une uvéite active malgré un traitement par corticoïdes (prednisone par voie orale à la dose de 10 à 60 mg/jour). Au moment de l'inclusion dans l'étude, tous les patients ont reçu une dose de 60 mg/jour de prednisone pendant deux semaines, progressivement réduite selon un schéma standardisé jusqu'à l'arrêt complet de la corticothérapie à la semaine 15.
L'étude UV II a évalué 226 patients présentant une uvéite inactive nécessitant une corticothérapie chronique (prednisone par voie orale à la dose de 10 à 35 mg/jour) au moment de l'inclusion dans l'étude pour contrôler leur maladie. La dose de corticoïdes était progressivement réduite selon un schéma standardisé jusqu'à l'arrêt complet de la corticothérapie à la semaine 19.
Le critère principal d'évaluation de l'efficacité dans les deux études était le « délai de survenue de la rechute ». La rechute était définie par un critère composite basé sur la présence de lésions vasculaires rétiniennes et/ou choriorétiniennes inflammatoires, le Tyndall cellulaire de la chambre antérieure, l'inflammation vitréenne et la meilleure acuité visuelle corrigée.
Les patients ayant terminé les études UV I et UV II étaient éligibles pour participer à une étude d'extension à long terme non contrôlée d'une durée initialement prévue de 78 semaines. Les patients ont été autorisés à continuer à prendre le médicament de l'étude au-delà de la semaine 78 jusqu'à ce qu'ils aient accès à l'adalimumab.
Réponse clinique
Les résultats des deux études ont mis en évidence une réduction statistiquement significative du risque de rechute chez les patients traités par l'adalimumab comparativement aux patients recevant le placebo (voir tableau 19). Les deux études ont montré un effet précoce et durable sur le taux de rechute sous adalimumab comparativement au placebo (voir figure 1).
Tableau 19 : Délai de survenue de la rechute dans les études UV I et UV II
| Analyse Traitement | N | Rechute N (%) | Délai médian de survenue de la rechute (mois) | HRa | IC à 95% pour le HRa | Valeur de pb |
| Délai de survenue de la rechute à la semaine 6 ou à partir de la semaine 6 dans l'étude UV I | ||||||
| Analyse principale (ITT) | | | | | | |
| Placebo | 107 | 84 (78,5) | 3,0 | - | - | - |
| Adalimumab | 110 | 60 (54,5) | 5,6 | 0,50 | 0,36 ; 0,70 | < 0,001 |
| Délai de survenue de la rechute à la semaine 2 ou à partir de la semaine 2 dans l'étude UV II | ||||||
| Analyse principale (ITT) | | | | | | |
| Placebo | 111 | 61 (55,0) | 8,3 | - | - | - |
| Adalimumab | 115 | 45 (39,1) | NEc | 0,57 | 0,39 ; 0,84 | 0,004 |
a HR pour l'adalimumab par rapport au placebo par une analyse de régression à risque proportionnel ajusté sur le traitement.
b Valeur de p bilatérale selon le test de log rank.
c NE = non estimable. Moins de la moitié des patients à risque ont présenté un événement.
Figure 1 : Courbes de Kaplan-Meier illustrant le délai de survenue de la rechute à la semaine 6 ou après la semaine 6 (étude UV I), ou à la semaine 2 ou après la semaine 2 (étude UV II)
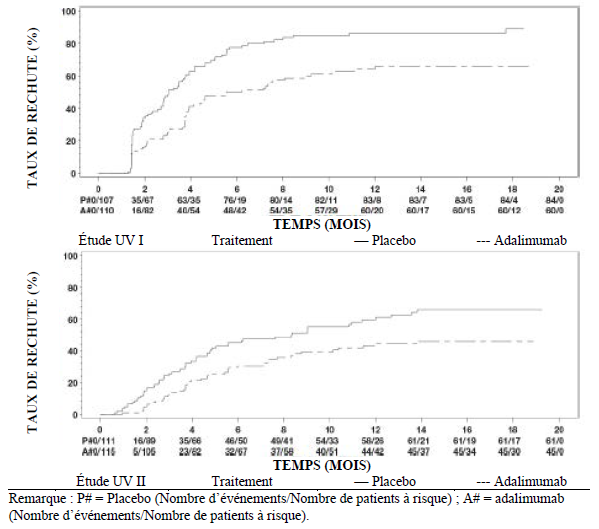
Dans l'étude UV I, des différences statistiquement significatives en faveur de l'adalimumab par rapport au placebo ont été observées pour chaque composante du critère de rechute. Dans l'étude UV II, des différences statistiquement significatives ont été observées pour l'acuité visuelle, toutes les autres composantes étaient cependant numériquement en faveur de l'adalimumab.
Sur les 424 patients inclus dans l'extension à long terme non contrôlée des études UV I et UV II, 60 patients ont été considérés inéligibles (par exemple, en raison de déviations ou en raison de complications secondaires à une rétinopathie diabétique, suite à une chirurgie de la cataracte ou une vitrectomie) et ont été exclus de l'analyse des critères primaires d'efficacité. Sur les 364 patients restants, 269 patients évaluables (74%) ont été traités pendant 78 semaines en ouvert par l'adalimumab. Sur la base des données observées, 216 (80,3 %) étaient en phase de quiescence(absence de lésions inflammatoires actives, Tyndall cellulaire ≤ 0,5+, inflammation du vitré de grade ≤ 0,5+) avec corticothérapie concomitante à une dose ≤ 7,5 mg par jour, et 178 (66,2 %) étaient en phase de quiescence sans corticoïdes. La meilleure acuité visuelle corrigée était soit améliorée soit maintenue (détérioration < 5 lettres) pour 88,6 % des yeux évalués à la semaine 78. Les données au- delà de la semaine 78 concordaient globalement avec ces résultats, mais le nombre de sujets inclus a diminué après cette période. Dans l'ensemble, parmi les patients sortis de l'étude, 18 % ont arrêté l'étude en raison d'événements indésirables et 8 % en raison d'une réponse insuffisante au traitement par l'adalimumab.
Qualité de vie
Les résultats rapportés par les patients en matière de qualité de vie liée à la fonction visuelle ont fait l'objet d'une évaluation dans les deux études cliniques, à l'aide du questionnaire de qualité de vie NEI VF-Q25. La majorité des sous-scores étaient numériquement en faveur de l'adalimumab, avec des différences moyennes statistiquement significatives en matière de vision générale, douleur oculaire, vision de près, santé mentale, et de score total dans l'étude UV I, et en matière de vision générale et santé mentale dans l'étude UV II. Les effets sur la qualité de vie liée à la fonction visuelle n'étaient pas numériquement en faveur de l'adalimumab pour le sous-score vision des couleurs dans l'étudeUV I et pour les sous-scores vision des couleurs, vision périphérique et vision de près dans l'étude UV II.
Immunogénicité
Pendant le traitement par l'adalimumab, des anticorps peuvent se développer contre ce principe actif. La formation d'anticorps anti-adalimumab est associée à une augmentation de la clairance et à une diminution de l'efficacité de l'adalimumab. Il n'y a pas de corrélation apparente entre la présence d'anticorps anti-adalimumab et la survenue d'effets indésirables.
Population pédiatrique
Arthrite juvénile idiopathique (AJI)
Arthrite juvénile idiopathique polyarticulaire (AJIp)
La tolérance et l'efficacité de l'adalimumab ont été évaluées dans deux études (AJIp I et II) chez des enfants ayant une arthrite juvénile idiopathique polyarticulaire active ou une AJI d'évolution polyarticulaire, qui présentaient différentes formes de début de la maladie (le plus souvent polyarthrite avec facteur rhumatoïde négatif ou positif et oligoarthrite étendue).
AJIp I
La tolérance et l'efficacité de l'adalimumab ont été évaluées dans une étude multicentrique randomisée en double aveugle en groupes parallèles menée chez 171 enfants (de 4 à 17 ans) présentant une AJI polyarticulaire. Dans la phase de pré-inclusion en ouvert (PI-O), les patients ont été répartis en deux groupes : patients traités par MTX (méthotrexate) ou non traités par MTX. Les patients de la strate « sans MTX » étaient naïfs de traitement ou le MTX avait été arrêté deux semaines au moins avant l'administration du médicament à l'étude. Les patients sont restés sous doses stables d'anti- inflammatoires non stéroïdiens (AINS) et/ou de prednisone (≤ 0,2 mg/kg/jour ou 10 mg/jour au maximum). Dans la phase de pré-inclusion en ouvert, tous les patients ont reçu 24 mg/m² de l'adalimumab, jusqu'à un maximum de 40 mg, toutes les deux semaines pendant 16 semaines. Le tableau 20 présente la distribution des patients par âge et doses minimales, médianes et maximales reçues pendant la phase de pré-inclusion en ouvert.
Tableau 20 : Distribution des patients par âge et doses d'adalimumab reçues pendant la phase de pré-inclusion en ouvert
| Groupe d'âge | Nombre de patients au début de l'étude n (%) | Dose minimale, médiane et maximale |
| 4 à 7 ans | 31 (18,1) | 10, 20 et 25 mg |
| 8 à 12 ans | 71 (41,5) | 20, 25 et 40 mg |
| 13 à 17 ans | 69 (40,4) | 25, 40 et 40 mg |
Les patients présentant une réponse ACR 30 pédiatrique en semaine 16 étaient éligibles pour être randomisés dans la phase en double aveugle (DA) et ils ont reçu l'adalimumab à raison de 24 mg/m² jusqu'à un maximum de 40 mg ou le placebo toutes les deux semaines pendant 32 semaines de plus ou jusqu'à une poussée de la maladie. Une poussée était définie comme une aggravation d'au moins 30% d'au moins 3 des 6 critères du score ACR pédiatrique, la présence d'au moins deux articulations actives et une amélioration supérieure à 30% d'un critère seulement sur les six. Après 32 semaines ou au moment d'une poussée de la maladie, les patients étaient éligibles pour entrer dans la phase d'extension en ouvert.
Tableau 21 : Réponses ACR 30 Pédiatrique dans l'étude de l'AJI
| Strate | MTX | Sans MTX | ||
| Phase | | | ||
| Pré-inclusion en ouvert de 16 semaines | | | ||
| Réponse ACR 30 Pédiatrique (n/N) | 94,1% (80/85) | 74,4% (64/86) | ||
| Critères d'efficacité | ||||
| Phase en double aveugle de 32 semaines | Adalimumab/MTX (N=38) | Placebo/MTX (N=37) | Adalimumab (N=30) | Placebo (N=28) |
| Poussées de la maladie à la fin des 32 semainesa (n/N) | 36,8% (14/38) | 64,9% (24/37)b | 43,3% (13/30) | 71,4% (20/28)c |
| Délai médian jusqu'à une poussée de la maladie | > 32 semaines | 20 semaines | > 32 semaines | 14 semaines |
b p = 0,015
c p = 0,031
Chez les sujets qui avaient répondu en semaine 16 (n = 144), les réponses ACR pédiatrique 30/50/70/90 ont été maintenues pendant des durées allant jusqu'à six ans dans la phase d'extension en ouvert chez les patients qui avaient reçu l'adalimumab pendant toute l'étude. Dix-neuf patients dont 11 du groupe d'âge de 4 à 12 ans et 8 du groupe d'âge de 13 à 17 ans ont été traités pendant 6 ans ou plus.
Les réponses globales ont été généralement supérieures et le nombre de patients ayant développé des anticorps a été plus faible avec l'association adalimumab plus MTX qu'avec l'adalimumab en monothérapie. En tenant compte de ces résultats, l'adalimumab est recommandé en association avec le MTX et en monothérapie chez les patients pour lesquels le traitement par MTX est inadapté (voir rubrique Posologie et mode d'administration).
AJIp II
La tolérance et l'efficacité de l'adalimumab ont été évaluées dans une étude en ouvert, multicentrique chez 32 enfants (âgés de 2 à 4 ans ou âgés de 4 ans et plus, d'un poids < 15 kg) présentant une AJI polyarticulaire modérément à sévèrement active. Les patients ont reçu l'adalimumab à la dose de 24 mg/m² de surface corporelle jusqu'à une dose maximale de 20 mg en une seule injection sous- cutanée toutes les 2 semaines pendant au moins 24 semaines. Durant l'étude, la plupart des patients avaient un traitement concomitant par MTX, une plus faible proportion avait des corticoïdes ou des AINS.
À la semaine 12 et à la semaine 24, la réponse ACR 30 Pédiatrique était de 93,5% et de 90,0%, respectivement, en utilisant les données observées. La proportion de patients avec ACR 50/70/90 Pédiatrique à la semaine 12 et à la semaine 24 était de 90,3%/61,3%/38,7% et 83,3%/73,3%/36,7%, respectivement. Parmi ceux ayant répondu (ACR 30 Pédiatrique) à la semaine 24 (n = 27 sur les30 patients), la réponse ACR 30 Pédiatrique était maintenue jusqu'à 60 semaines dans la phase d'extension en ouvert chez les patients qui ont reçu l'adalimumab durant toute cette période. Globalement, 20 patients ont été traités pendant 60 semaines ou plus.
Arthrite liée à l'enthésite
La tolérance et l'efficacité de l'adalimumab ont été évaluées dans une étude multicentrique, randomisée en double aveugle chez 46 patients pédiatriques (âgés de 6 à 17 ans) présentant une arthrite liée à l'enthésite modérée. Les patients ont été randomisés pour recevoir, toutes les deux semaines pendant 12 semaines, soit une dose d'adalimumab de 24 mg/m² de surface corporelle jusqu'à une dose maximale de 40 mg, soit un placebo. La phase en double aveugle a été suivie d'une phase en ouvert durant laquelle les patients recevaient une dose d'adalimumab de 24 mg/m² de surface corporelle jusqu'à une dose maximale de 40 mg toutes les deux semaines par voie sous-cutanée pendant 192 semaines supplémentaires. Le critère d'évaluation principal était la variation en pourcentage du nombre d'articulations actives touchées par l'arthrite (gonflement non lié à une malformation ou articulations avec perte de mouvement et douleur et/ou sensibilité) entre l'inclusion et la semaine 12. Une réduction moyenne de -62,6% (variation médiane : -88,9%) a été observée chez les patients traités par l'adalimumab par rapport à -11,6% (variation médiane : -50,0%) chez les patients recevant le placebo. L'amélioration relative au nombre d'articulations actives touchées par l'arthrite a été maintenue au cours de la phase en ouvert jusqu'à la semaine 156 pour les 26 patients sur les 31 (84%) du groupe traités par l'adalimumab qui sont restés dans l'étude. Une amélioration clinique mais non statistiquement significative a été observée chez la majorité des patients pour les critères secondaires tels que le nombre de sites d'enthésite, le nombre d'articulations douloureuses, le nombre d'articulations gonflées, et la réponse selon les critères ACR 50 et 70 pédiatriques.
Psoriasis en plaques chez l'enfant et l'adolescent
L'efficacité de l'adalimumab a été évaluée dans une étude contrôlée, randomisée en double aveugle chez 114 patients pédiatriques âgés de 4 ans et plus présentant un psoriasis en plaques chronique sévère (défini par un score PGA (Physician's Global Assessment) ≥ 4 ou une atteinte de la surface corporelle > 20% ou > 10% avec des lésions très épaisses ou un score PASI (Psoriasis Area and Severity Index) ≥ 20 ou ≥ 10 avec atteinte cliniquement significative du visage, des organes génitaux ou des mains et/ou pieds) qui n'était pas suffisamment contrôlé par un traitement topique et l'héliothérapie ou la photothérapie.
Les patients ont reçu l'adalimumab à la dose de 0,8 mg/kg toutes les deux semaines (jusqu'à 40 mg), ou 0,4 mg/kg toutes les deux semaines (jusqu'à 20 mg) ou le méthotrexate à la dose de 0,1 à 0,4 mg/kg une fois par semaine (jusqu'à 25 mg). À la semaine 16, il y a eu plus de répondeurs (par ex. PASI 75) chez les patients randomisés dans le groupe de l'adalimumab 0,8 mg/kg toutes les deux semaines par rapport aux patients randomisés dans le groupe de l'adalimumab 0,4 mg/kg toutes les deux semaines ou le MTX.
Tableau 22 : Psoriasis en plaques pédiatrique - Résultats d'efficacité à 16 semaines
| | MTXa N=37 | Adalimumab 0,8 mg/kg toutes les deux semaines N = 38 |
| PASI 75b | 12 (32,4%) | 22 (57,9%) |
| PGA : clair/minimalc | 15 (40,5%) | 23 (60,5%) |
b p = 0,027, adalimumab 0,8 mg/kg versus MTX
c p = 0,083, adalimumab 0,8 mg/kg versus MTX
Chez les patients ayant obtenu un score PASI 75 et un score PGA « clair ou minimal », le traitement a été arrêté pendant une durée allant jusqu'à 36 semaines et ils ont été suivis pour détecter une perte de contrôle de la maladie (c'est-à-dire une aggravation d'au moins 2 grades du score PGA). Les patients ont ensuite été retraités par l'adalimumab 0,8 mg/kg toutes les deux semaines pendant 16 semaines supplémentaires et les taux de réponse observés pendant le retraitement ont été comparables à ceux rapportés pendant la phase en double aveugle antérieure : réponse PASI 75 chez 78,9% des patients (15 sur 19) et score PGA « clair ou minimal » chez 52,6% (10 sur 19).
Dans la phase en ouvert de l'étude, les réponses PASI 75 et PGA « clair ou minimal » ont été maintenues pendant une durée allant jusqu'à 52 semaines supplémentaires sans nouveaux signaux de sécurité.
Hidrosadénite suppurée chez l'adolescent
Il n'existe pas d'essai clinique conduit avec l'adalimumab chez des adolescents atteints d'hidrosadénite suppurée. L'efficacité de l'adalimumab pour le traitement des adolescents atteints d'HS est prédite sur la base de l'efficacité et de la relation exposition-réponse démontrées chez des patients adultes atteints d'HS ainsi que de la probabilité que l'évolution de la maladie, sa physiopathologie et les effets du médicament soient sensiblement similaires à ceux observés chez les adultes aux mêmes niveaux d'exposition. La tolérance de la dose recommandée d'adalimumab chez les adolescents atteints d'HS est basée sur le profil de tolérance de l'adalimumab dans ses indications croisées chez les adultes et chez les patients pédiatriques à des doses similaires ou plus fréquentes (voir rubrique Propriétés pharmacocinétiques).
Maladie de Crohn de l'enfant et l'adolescent
Une étude clinique multicentrique, randomisée, en double aveugle a évalué l'efficacité et la tolérance de l'adalimumab dans le traitement d'induction et le traitement d'entretien à des doses déterminées en fonction du poids (< 40 kg ou ≥ 40 kg) chez 192 enfants et adolescents âgés de 6 à 17 ans (inclus), présentant une maladie de Crohn (MC) modérée à sévère (définie par un indice d'activité de la maladie de Crohn chez l'enfant (PCDAI [Paediatric Crohn's Disease Activity Index]) > 30). Les patients devaient ne pas avoir répondu à un traitement conventionnel de la MC (comprenant un corticoïde et/ou un immunomodulateur). Les patients pouvaient également ne plus répondre ou être intolérants à l'infliximab.
Tous les patients ont reçu un traitement d'induction en ouvert à une dose déterminée en fonction de leur poids initial : 160 mg à la semaine 0 et 80 mg à la semaine 2 pour les patients dont le poids est ≥ 40 kg, et respectivement 80 mg et 40 mg pour les patients dont le poids est < 40 kg.
À la semaine 4, les patients ont été randomisés selon un rapport 1/1, en fonction de leur poids à cette date, pour recevoir le schéma posologique d'entretien soit à dose faible soit à dose standard, comme le montre le tableau 23.
Tableau 23 : Schéma posologique d'entretien
| Poids du patient | Dose faible | Dose standard |
| < 40 kg | 10 mg toutes les deux semaines | 20 mg toutes les deux semaines |
| ≥40 kg | 20 mg toutes les deux semaines | 40 mg toutes les deux semaines |
Résultats d'efficacité
Le critère de jugement principal de l'étude était la rémission clinique à la semaine 26, définie par un score PCDAI ≤ 10.
Les taux de rémission clinique et de réponse clinique (définie par une réduction du score PCDAI d'au moins 15 points par rapport à la valeur initiale) sont présentés dans le tableau 24. Les taux d'arrêt des corticoïdes ou des immunomodulateurs sont présentés dans le tableau 25.
Tableau 24 : Étude sur la MC pédiatrique - Rémission et réponse clinique (PCDAI)
| | Dose standard 40/20 mg toutes les deux semaines N = 93 | Dose faible 20/10 mg toutes les deux semaines N = 95 | Valeur de p* |
| Semaine 26 | | | |
| Rémission clinique | 38,7% | 28,4% | 0,075 |
| Réponse clinique | 59,1% | 48,4% | 0,073 |
| Semaine 52 | | | |
| Rémission clinique | 33,3% | 23,2% | 0,100 |
| Réponse clinique | 41,9% | 28,4% | 0,038 |
Tableau 25 : Étude sur la MC pédiatrique - Arrêt des corticoïdes ou des immunomodulateurs et fermeture des fistules
| | Dose standard 40/20 mg toutes les deux semaines | Dose faible 20/10 mg toutes les deux semaines | Valeur de p1 |
| Arrêt des corticoïdes | N = 33 | N = 38 | |
| Semaine 26 | 84,8% | 65,8% | 0,066 |
| Semaine 52 | 69,7% | 60,5% | 0,420 |
| Arrêt des immunomodulateurs2 | N = 60 | N = 57 | |
| Semaine 52 | 30,0% | 29,8% | 0,983 |
| Fermeture des fistules3 | N = 15 | N = 21 | |
| Semaine 26 | 46,7% | 38,1% | 0,608 |
| Semaine 52 | 40,0% | 23,8% | 0,303 |
2 Le traitement immunosuppresseur ne pouvait être arrêté qu'à partir de la semaine 26, à la libre appréciation de l'investigateur, si le patient répondait au critère de réponse clinique
3 Définie comme la fermeture de toutes les fistules drainantes, au moins 2 visites consécutives après la visite initiale.
Des augmentations statistiquement significatives (amélioration) de l'indice de masse corporelle et de la vitesse de croissance staturale ont été observées dans les deux groupes de traitement entre la visite initiale et les semaines 26 et 52.
Des améliorations statistiquement et cliniquement significatives par rapport à la visite initiale ont également été observées dans les deux groupes de traitement pour les paramètres de qualité de vie (y compris IMPACT III).
Cent patients (n = 100) issus de l'étude sur la MC pédiatrique ont été inclus dans une étude d'extension à long terme, en ouvert. Après 5 ans de traitement par l'adalimumab, 74,0% (37/50) des 50 patients restant dans l'étude continuaient à être en rémission clinique et 92,0% (46/50) des patients continuaient à être en réponse clinique selon le score PCDAI.
Rectocolite hémorragique chez l'enfant et l'adolescent
La tolérance et l'efficacité de l'adalimumab ont été évaluées dans une étude multicentrique, randomisée, en double aveugle, menée chez 93 patients âgés de 5 à 17 ans et atteints de rectocolite hémorragique modérée à sévère (score Mayo de 6 à 12 avec un sous-score endoscopique de 2 à 3 points, confirmé par une endoscopie évaluée par une relecture centralisée) qui présentaient une réponse inadéquate ou une intolérance au traitement conventionnel. Environ 16 % des patients del'étude étaient en échec d'un précédent traitement par anti -TNF. Les patients recevant des corticoïdes au moment de l'inclusion ont été autorisés à diminuer progressivement leur traitement par corticoïdes après la semaine 4.
Au cours de la période d'induction de l'étude, 77 patients ont été randomisés (3:2) pour recevoir un traitement en double aveugle par adalimumab à une dose d'induction de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0 et à la semaine 1, et de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 ; ou une dose d'induction de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0, un placebo à la semaine 1 et une dose de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2. Les deux groupes ont reçu une dose de 0,6 mg/kg (dose maximale de 40 mg) à la semaine 4 et à la semaine 6. Suite à un amendement de l'étude, 16 patients ont reçu un traitement en ouvert par adalimumab à la dose d'induction de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0 et à la semaine 1, et de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2.
À la semaine 8, 62 patients ayant présenté une réponse clinique selon le Score Mayo partiel (SMP ; définie comme une diminution du SMP ≥ 2 points et ≥ 30 % par rapport à la valeur initiale) ont été randomisés pour recevoir un traitement d'entretien en double aveugle par adalimumab à une dose de 0,6 mg/kg (dose maximale de 40 mg) chaque semaine, ou une dose d'entretien de 0,6 mg/kg (dose maximale de 40 mg) toutes les deux semaines. Après l'amendement, 12 patients ayant présenté une réponse clinique d'après le SMP ont été randomisés pour recevoir un placebo, mais ces derniers n'ont pas été inclus dans l'analyse principale d'efficacité.
Une poussée de la maladie était définie comme une augmentation du SMP d'au moins 3 points (pour les patients présentant un SMP de 0 à 2 à la semaine 8), d'au moins 2 points (pour les patients présentant un SMP de 3 à 4 à la semaine 8), ou d'au moins 1 point (pour les patients présentant un SMP de 5 à 6 à la semaine 8).
Les patients dont l'état répondait aux critères de poussée de la maladie à la semaine 12 ou après ont été randomisés pour recevoir une dose de réinduction de 2,4 mg/kg (dose maximale de 160 mg) ou une dose de 0,6 mg/kg (dose maximale de 40 mg) et ont conservé leur dose d'entretien respective par la suite.
Résultats d'efficacité
Les co-critères principaux d'évaluation de l'étude étaient la rémission clinique selon le SMP (définie par un SMP ≤ 2 sans aucun sous-score individuel > 1) à la semaine 8, et la rémission clinique selon le score SMT (score Mayo total) (définie par un score Mayo ≤ 2 sans aucun sous-score individuel > 1) à la semaine 52 chez les patients ayant obtenu une réponse clinique d'après le SMP à la semaine 8.
À la semaine 8, les taux de rémission clinique selon le SMP pour les patients de chaque groupe ayant reçu le traitement d'induction par adalimumab en double aveugle sont présentés dans le tableau 26.
Tableau 26 : Rémission clinique selon le SMP à 8 semaines
| | Adalimumaba Dose maximale de 160 mg à la semaine 0 / placebo à la semaine 1 N = 30 | Adalimumabb,c Dose maximale de 160 mg à la semaine 0 et à la semaine 1 N = 47 |
| Rémission clinique | 13/30 (43,3 %) | 28/47 (59,6 %) |
| a
Adalimumab 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0, placebo
à la semaine 1, et 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 b Adalimumab 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0 et à la semaine 1, et 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 c En excluant la dose d'induction en ouvert d'adalimumab de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0 et à la semaine 1, et de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 Remarque 1 : les deux groupes du traitement d'induction ont reçu une dose de 0,6 mg/kg (dose maximale de 40 mg) à la semaine 4 et à la semaine 6. Remarque 2 : pour les patients dont les valeurs étaient manquantes à la semaine 8, le critère d'évaluation est considéré comme n'étant pas atteint. | ||
À la semaine 52, la rémission clinique selon le SMT chez les patients ayant présenté une réponse clinique à la semaine 8, la réponse clinique selon le SMT (définie comme une diminution du score Mayo ≥ 3 points et ≥ 30 %) chez les patients ayant présenté une réponse à la semaine 8, la cicatrisation de la muqueuse selon le SMT (définie comme un score endoscopique Mayo ≤ 1) chez les patients ayant présenté une réponse à la semaine 8, la rémission clinique selon le SMT chez les patients en rémission à la semaine 8, et la proportion de patients en rémission sans corticoïdes selon le SMT chez les patients ayant présenté une réponse à la semaine 8 ont été évaluées pour les patients ayant reçu l'adalimumab en double aveugle à la dose d'entretien maximale de 40 mg 1 semaine sur 2 (0,6 mg/kg) et à la dose d'entretien maximale de 40 mg chaque semaine (0,6 mg/kg) (tableau 27).
Tableau 27 : Résultats d'efficacité à 52 semaines
| | Adalimumaba Dose maximale de 40 mg 1 semaine sur 2 N = 31 | Adalimumabb Dose maximale de 40 mg chaque semaine N = 31 |
| Rémission clinique chez les répondeurs à la semaine 8 selon le SMP | 9/31 (29,0 %) | 14/31 (45,2 %) |
| Réponse clinique chez les répondeurs à la semaine 8 selon le SMP | 19/31 (61,3 %) | 21/31 (67,7 %) |
| Cicatrisation de la muqueuse chez les répondeurs à la semaine 8 selon le SMP | 12/31 (38,7 %) | 16/31 (51,6 %) |
| Rémission clinique chez les patients en rémission à la semaine 8 selon le SMP | 9/21 (42,9 %) | 10/22 (45,5 %) |
| Rémission sans corticoïdes chez les répondeurs à la semaine 8 selon le SMPc | 4/13 (30,8 %) | 5/16 (31,3 %) |
| a Adalimumab 0,6 mg/kg (dose maximale de 40 mg) toutes les deux semaines b Adalimumab 0,6 mg/kg (dose maximale de 40 mg) chaque semaine c Pour les patients recevant un traitement concomitant par corticoïdes à l'inclusion. Remarque : les patients dont les valeurs étaient manquantes à la semaine 52 ou qui ont été randomisés pour recevoir un traitement de réinduction ou d'entretien ont été considérés comme non répondeurs d'après les critères d'évaluation de la semaine 52. | ||
Les autres critères d'évaluation exploratoires de l'efficacité comprenaient la réponse clinique selon l'indice d'activité de la rectocolite hémorragique pédiatrique (Paediatric Ulcerative Colitis Activity Index, PUCAI) (définie comme une diminution de l'indice PUCAI ≥ 20 points par rapport à la valeur initiale) et la rémission clinique selon l'indice PUCAI (définie comme un indice PUCAI < 10) à la semaine 8 et à la semaine 52 (tableau 28).
Tableau 28 : Résultats des critères d'évaluation exploratoires selon l'indice PUCAI
| | Semaine 8 | |
| Adalimumaba Dose maximale de 160 mg à la semaine 0 / placebo à la semaine 1 N = 30 | Adalimumabb,c Dose maximale de 160 mg à la semaine 0 et à la semaine 1 N = 47 | |
| Rémission clinique selon l'indice PUCAI | 10/30 (33,3 %) | 22/47 (46,8 %) |
| Réponse clinique selon l'indice PUCAI | 15/30 (50,0 %) | 32/47 (68,1 %) |
| | Semaine 52 | |
| Adalimumabd Dose maximale de 40 mg 1 semaine sur 2 N = 31 | Adalimumabe Dose maximale de 40 mg chaque semaine N = 31 | |
| Rémission clinique chez les répondeurs à la semaine 8 selon l'indice PUCAI | 14/31 (45,2 %) | 18/31 (58,1 %) |
| Réponse clinique chez les répondeurs à la semaine 8 selon l'indice PUCAI | 18/31 (58,1 %) | 16/31 (51,6 %) |
| a
Adalimumab 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0, placebo
à la semaine 1, et 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 b Adalimumab 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0 et à la semaine 1, et 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 c En excluant la dose d'induction en ouvert d'adalimumab de 2,4 mg/kg (dose maximale de 160 mg) à la semaine 0 et à la semaine 1, et de 1,2 mg/kg (dose maximale de 80 mg) à la semaine 2 d Adalimumab 0,6 mg/kg (dose maximale de 40 mg) toutes les deux semaines e Adalimumab 0,6 mg/kg (dose maximale de 40 mg) chaque semaine Remarque 1 : les deux groupes du traitement d'induction ont reçu une dose de 0,6 mg/kg (dose maximale de 40 mg) à la semaine 4 et à la semaine 6. Remarque 2 : pour les patients dont les valeurs étaient manquantes à la semaine 8, les critères d'évaluation sont considérés comme n'étant pas atteints. Remarque 3 : les patients dont les valeurs étaient manquantes à la semaine 52 ou qui ont été randomisés pour recevoir un traitement de réinduction ou d'entretien ont été considérés comme non répondeurs d'après les critères d'évaluation de la semaine 52. | ||
Parmi les patients traités par adalimumab qui ont reçu un traitement de réinduction pendant la période d'entretien, 2/6 (33 %) ont obtenu une réponse clinique selon le SMT à la semaine 52.
Qualité de vie
Par rapport aux données d'inclusion, des améliorations cliniquement significatives ont été observées au niveau des paramètres IMPACT III et des scores du questionnaire à remplir par le soignant sur la productivité au travail et la limitation des activités (Work Productivity and Activity Impairment, WPAI) dans les groupes traités par adalimumab.
Par rapport aux données d'inclusion, des augmentations cliniquement significatives (amélioration) de la vitesse de croissance staturale ont été observées dans les groupes traités par adalimumab, et des augmentations cliniquement significatives (amélioration) de l'indice de masse corporelle ont été observées chez les sujets recevant la dose d'entretien élevée de 40 mg (0,6 mg/kg) maximum chaque semaine.
Uvéite chez l'enfant et l'adolescent
La tolérance et l'efficacité de l'adalimumab ont été évaluées dans une étude randomisée, contrôlée, en double aveugle, conduite chez 90 patients pédiatriques âgés de 2 à < 18 ans atteints d'uvéite active antérieure non infectieuse associée à une AJI qui étaient réfractaires à un traitement d'au moins 12 semaines par le méthotrexate. Les patients ont reçu soit un placebo soit 20 mg d'adalimumab (s'ils pesaient < 30 kg) ou 40 mg d'adalimumab (s'ils pesaient ≥ 30 kg) toutes les deux semaines en association avec leur dose initiale de méthotrexate.
Le critère principal d'évaluation était le « délai de survenue de la rechute ». Les critères déterminant la rechute étaient une aggravation ou l'absence prolongée d'amélioration de l'inflammation oculaire, une amélioration partielle avec le développement de comorbidités oculaires prolongées ou l'aggravation des comorbidités oculaires, l'utilisation non autorisée de médicaments concomitants et la suspension du traitement sur une durée de temps prolongée.
Réponse clinique
L'adalimumab a retardé de manière significative le délai de survenue de la rechute par rapport au placebo (voir figure 2, p < 0,0001, test de log rank). Le délai médian de survenue de la rechute était de 24,1 semaines pour les patients recevant le placebo, tandis que le délai médian de survenue de la rechute n'a pas pu être estimé pour les patients traités par l'adalimumab car moins de la moitié de ces patients a présenté une rechute. L'adalimumab a diminué de manière significative le risque de rechute de 75% par rapport au placebo, comme le montre le « hazard ratio » (HR = 0,25 [IC à 95% : 0,12 ; 0,49]).
Figure 2 : Courbes de Kaplan-Meier illustrant le délai de survenue de la rechute dans l'étude sur l'uvéite pédiatrique
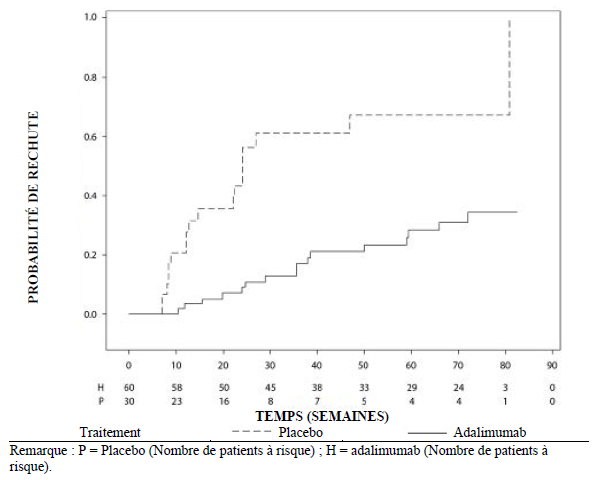
Absorption et distribution
Après administration sous-cutanée de 24 mg/m² (une dose maximale de 40 mg) toutes les deux semaines chez des patients atteints d'arthrite juvénile idiopathique (AJI) polyarticulaire âgés de 4 à 17 ans, la concentration sérique résiduelle moyenne de l'adalimumab (valeurs mesurées des semaines 20 à 48) a été de 5,6 ± 5,6 µg/mL (CV 102%) lorsque l'adalimumab était administré sans traitement concomitant par le méthotrexate et de 10,9 ± 5,2 µg/mL (CV 47,7%) en cas d'administration concomitante avec le méthotrexate.
Chez les patients atteints d'AJI polyarticulaire âgés de 2 à < 4 ans ou âgés de 4 ans et plus d'un poids < 15 kg ayant reçu l'adalimumab à la dose de 24 mg/m², la concentration sérique résiduelle moyenne de l'adalimumab était de 6,0 ± 6,1 µg/mL (CV 101%) lorsque l'adalimumab était administré sans traitement concomitant par le méthotrexate et de 7,9 ± 5,6 µg/mL (CV 71,2%) en cas d'administration concomitante avec le méthotrexate.
Après administration sous-cutanée d'une dose de 24 mg/m² (une dose maximale de 40 mg) toutes les deux semaines chez des patients atteints d'arthrite liée à l'enthésite âgés de 6 à 17 ans, la concentration sérique résiduelle moyenne de l'adalimumab à l'état d'équilibre (valeurs mesurées à la semaine 24) a été de 8,8 ± 6,6 µg/mL lorsque l'adalimumab était administré sans traitement concomitant par le méthotrexate et de 11,8 ± 4,3 µg/mL en cas d'administration concomitante avec le méthotrexate.
Après administration sous-cutanée de 0,8 mg/kg (une dose maximale de 40 mg) toutes les deux semaines chez des patients pédiatriques atteints de psoriasis en plaques chronique, la concentration résiduelle moyenne à l'état d'équilibre de l'adalimumab (moyenne ± ET) était d'environ 7,4 ± 5,8 µg/mL (CV 79%).
L'exposition à l'adalimumab chez les adolescents atteints d'HS a été prédite en utilisant une modélisation pharmacocinétique de population et une simulation basées sur la pharmacocinétique observée dans des indications croisées chez d'autres patients pédiatriques (psoriasis pédiatrique, arthrite juvénile idiopathique, maladie de Crohn pédiatrique et arthrite liée à l'enthésite). Le schéma posologique recommandé d'Hulio chez les adolescents atteints d'hidrosadénite suppurée est 40 mg toutes les deux semaines. Comme l'exposition à l'adalimumab peut être modifiée par la masse corporelle, les adolescents avec un poids corporel plus élevé et une réponse insuffisante pourraient bénéficier de la dose recommandée chez l'adulte de 40 mg par semaine.
Chez les enfants et les adolescents atteints de MC modérée à sévère, la dose d'induction de l'adalimumab en ouvert était respectivement de 160/80 mg ou 80/40 mg aux semaines 0 et 2, en fonction d'une valeur seuil de poids de 40 kg. À la semaine 4, les patients ont été randomisés selon un rapport de 1/1 pour recevoir un traitement d'entretien soit à la dose standard (40/20 mg toutes les deux semaines), soit à la dose faible (20/10 mg toutes les deux semaines) en fonction de leur poids. Les concentrations sériques résiduelles moyennes (± ET) de l'adalimumab obtenues à la semaine 4 ont été de 15,7 ± 6,6 µg/mL chez les patients d'un poids ≥ 40 kg (160/80 mg) et de 10,6 ± 6,1 µg/mL chez les patients d'un poids < 40 kg (80/40 mg).
Chez les patients toujours traités, les concentrations résiduelles moyennes (± ET) de l'adalimumab à la semaine 52 étaient de 9,5 ± 5,6 µg/mL dans le groupe traité à la dose standard et de 3,5 ± 2,2 µg/mL dans le groupe traité à la dose faible. Les concentrations résiduelles moyennes ont été maintenues chez les patients ayant continué à recevoir le traitement par l'adalimumab toutes les deux semaines pendant 52 semaines. Chez les patients dont le schéma posologique est passé de toutes les deux semaines à toutes les semaines, les concentrations sériques moyennes (± ET) de l'adalimumab à la semaine 52 ont été de 15,3 ± 11,4 µg/mL (40/20 mg, toutes les semaines) et de 6,7 ± 3,5 µg/mL (20/10 mg, toutes les semaines).
L'exposition à l'adalimumab chez les patients atteints d'uvéite pédiatrique a été prédite en utilisant une modélisation pharmacocinétique de population et une simulation basée sur la pharmacocinétique observée dans différentes indications pédiatriques (psoriasis pédiatrique, arthrite juvénile idiopathique, maladie de Crohn pédiatrique et arthrite liée à l'enthésite). Aucune donnée d'exposition clinique n'est disponible sur l'utilisation d'une dose de charge chez les enfants âgés de moins de 6 ans. Les expositions prévisibles indiquent qu'en l'absence de méthotrexate, une dose de charge peut entraîner une augmentation initiale de l'exposition systémique.
Relation exposition-réponse dans la population pédiatrique
Sur la base des données des essais cliniques chez les patients atteints d'AJI (AJI polyarticulaire et arthrite liée à l'enthésite), une relation exposition-réponse a été démontrée entre les concentrations plasmatiques et la réponse ACR Péd. 50. La concentration plasmatique d'adalimumab apparente produisant la moitié de la probabilité maximale de réponse ACR Péd. 50 (CE50) était de 3 µg/mL (IC à 95% : 1 - 6 µg/mL).
Des relations exposition-réponse entre la concentration d'adalimumab et l'efficacité chez les patients pédiatriques atteints de psoriasis en plaques sévère ont été établies pour les résultats PASI 75 et PGA « clair ou minimal », respectivement. Les taux de résultats PASI 75 et PGA « clair ou minimal » ont augmenté à mesure de l'augmentation des concentrations d'adalimumab, avec une CE50 apparente similaire d'environ 4,5 µg/mL (IC à 95% respectifs de 0,4 - 47,6 et 1,9 - 10,5) dans les deux cas.
Adultes
Après administration sous-cutanée d'une dose unique de 40 mg, l'absorption et la distribution de l'adalimumab ont été lentes, le pic de concentration sérique étant atteint 5 jours environ après l'administration. La biodisponibilité absolue moyenne de l'adalimumab, estimée à partir de trois études, a été de 64% après une dose sous-cutanée unique de 40 mg. Après administration de doses intraveineuses uniques variant de 0,25 à 10 mg/kg, les concentrations ont été proportionnelles à la dose. Après administration de doses de 0,5 mg/kg (~ 40 mg), les clairances étaient de 11 à 15 mL/heure, le volume de distribution (Vss) était compris entre 5 et 6 litres, la demi-vie terminale moyenne a été de deux semaines environ. La concentration d'adalimumab dans le liquide synovial de plusieurs patients atteints de polyarthrite rhumatoïde était comprise entre 31 et 96% des concentrations sériques.
Après administration sous-cutanée de 40 mg d'adalimumab toutes les deux semaines chez des patients adultes atteints de polyarthrite rhumatoïde (PR), les concentrations moyennes au creux étaient de l'ordre d'environ 5 µg/mL (sans méthotrexate) et de 8 à 9 µg/mL (avec méthotrexate). Les concentrations sériques minimales d'adalimumab à l'état d'équilibre ont augmenté de façon à peu près dose-dépendante après l'administration par voie sous-cutanée de 20, 40 et 80 mg toutes les deux semaines et toutes les semaines.
Chez les patients adultes atteints de psoriasis, la concentration minimale moyenne à l'état d'équilibre était de 5 µg/mL pendant le traitement par 40 mg d'adalimumab une semaine sur deux en monothérapie.
Chez les patients adultes atteints d'hidrosadénite suppurée, une dose de 160 mg d'adalimumab à la semaine 0 suivie d'une dose de 80 mg à la semaine 2, ont permis d'obtenir des concentrations sériques résiduelles d'adalimumab d'environ 7 à 8 µg/mL à la semaine 2 et à la semaine 4. Au cours du traitement par l'adalimumab 40 mg par semaine, les concentrations sériques résiduelles moyennes à l'état d'équilibre, de la semaine 12 jusqu'à la semaine 36, ont été d'environ 8 à 10 µg/mL.
Chez les patients atteints de la maladie de Crohn, la dose de charge de 80 mg d'adalimumab à la semaine 0 suivie de 40 mg d'adalimumab à la semaine 2 permet d'obtenir des concentrations sériques minimales d'adalimumab d'environ 5,5 µg/mL pendant la période d'induction. Une dose de charge de 160 mg d'adalimumab à la semaine 0 suivie de 80 mg d'adalimumab à la semaine 2 permet d'obtenir des concentrations sériques minimales d'adalimumab d'environ 12 µg/mL pendant la période d'induction. Des concentrations minimales moyennes à l'état d'équilibre d'environ 7 µg/mL ont été obtenues chez des patients atteints de la maladie de Crohn ayant reçu une dose d'entretien de 40 mg d'adalimumab toutes les deux semaines.
Après administration sous-cutanée de la dose déterminée par le poids de 0,6 mg/kg (dose maximale de 40 mg) toutes les deux semaines chez des enfants et des adolescents atteints de rectocolite hémorragique, la concentration sérique résiduelle moyenne de l'adalimumab à l'état d'équilibre était de 5,01 ± 3,28 µg/mL à la semaine 52. Chez les patients qui recevaient une dose de 0,6 mg/kg (dose maximale de 40 mg) chaque semaine, la concentration sérique résiduelle moyenne (± ET) de l'adalimumab à l'état d'équilibre était de 15,7 ± 5,60 µg/mL à la semaine 52.
Chez les patients adultes atteints d'uvéite, la dose d'induction de 80 mg d'adalimumab à la semaine 0 suivie de 40 mg d'adalimumab toutes les deux semaines à partir de la semaine 1 a permis d'obtenir des concentrations sériques moyennes d'adalimumab à l'état d'équilibre d'environ 8 à 10 µg/mL.
Une
modélisation et une simulation pharmacocinétiques et
pharmacocinétiques/pharmacodynamiques de population ont prédit une
exposition et une efficacité comparables chez les patients traités par
80 mg toutes les deux semaines en comparaison avec 40 mg toutes les
semaines (y compris les patients adultes atteints de PR, HS, RCH, MC ou
Ps, les adolescents atteints d'HS, et les patients pédiatriques≥ 40 kg atteints de MC et RCH).
Élimination
Les analyses pharmacocinétiques de populations portant sur des données recueillies chez plus de 1 300 patients atteints de PR, ont révélé une tendance à une augmentation de la clairance apparente de l'adalimumab avec une augmentation du poids corporel. Après ajustement en fonction des différences pondérales, le sexe et l'âge ont semblé avoir peu d'effet sur la clairance de l'adalimumab. Il a été observé que les taux sériques d'adalimumab libre (non lié aux anticorps anti-adalimumab, AAA) étaient plus bas chez les patients dont les AAA étaient mesurables.
Insuffisance hépatique ou rénale
L'adalimumab n'a pas été étudié chez les patients présentant une insuffisance hépatique ou rénale.
Hulio peut avoir une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Des vertiges et des troubles visuels peuvent survenir après l'administration d'Hulio (voir rubrique Effets indésirables).
Les données non cliniques issues des études de toxicologie en
administration unique, toxicologie en administration répétée et de
génotoxicité, n'ont pas révélé de risque particulier pour l'homme.
Une étude de toxicité portant sur le développement embryo-fœtal et le développement périnatal a été réalisée chez des singes cynomolgus à 0,30 et 100 mg/kg (9 à 17 singes/groupe) ; elle n'a pas révélé de signe de fœto-toxicité de l'adalimumab. Ni une étude du pouvoir carcinogène, ni une évaluation standard sur la fertilité et la toxicité post-natale n'ont été effectuées avec l'adalimumab en raison du manque de modèles appropriés pour un anticorps présentant une réactivité croisée limitée avec le TNF de rongeur et du développement d'anticorps neutralisants chez le rongeur.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Prescription réservée aux spécialistes et services en
DERMATOLOGIE
Prescription réservée aux spécialistes et services en
HEPATO/GASTRO-ENTEROLOGIE
Prescription réservée aux spécialistes et services
en MEDECINE INTERNE
Prescription réservée aux spécialistes et services en
OPHTALMOLOGIE
Prescription réservée aux spécialistes et services en
PEDIATRIE
Prescription réservée aux spécialistes et services en RHUMATOLOGIE
Médicament d'exception.
Remboursement en fonction de l'indication (JO du 20/12/2018) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont les suivantes :
- Arthrite juvénile idiopathique :
. Arthrite juvénile idiopathique polyarticulaire en association au méthotrexate : traitement de l'arthrite juvénile idiopathique polyarticulaire évolutive chez les patients à partir de 2 ans en cas de réponse insuffisante à un ou plusieurs traitements de fond. HULIO peut être administré en monothérapie en cas d'intolérance au méthotrexate ou lorsque la poursuite du traitement par le méthotrexate est inadaptée. L'adalimumab n'a pas été étudié chez les patients de moins de 2 ans.
. Arthrite liée à l'enthésite : traitement de l'arthrite active liée à l'enthésite chez les patients à partir de 6 ans en cas de réponse insuffisante ou d'intolérance au traitement conventionnel.
- Psoriasis en plaques de l'enfant et l'adolescent : traitement du psoriasis en plaques de l'enfant à partir de 4 ans et de l'adolescent, chez les patients ayant un psoriasis en plaques chronique sévère défini par :
. un échec (réponse insuffisante, contre-indication ou intolérance) à au moins deux traitements parmi les traitements systémiques non biologiques et la photothérapie ;
. et une forme étendue et/ou un retentissement psychosocial important.
- Maladie de Crohn chez l'enfant et l'adolescent : traitement de la maladie de Crohn active sévère, chez les enfants et les adolescents à partir de 6 ans, qui n'ont pas répondu à un traitement conventionnel comprenant un traitement nutritionnel de première intention, un corticoïde et un immunomodulateur, ou chez lesquels ces traitements sont mal tolérés ou contre-indiqués.
- Uvéite chez l'enfant et l'adolescent en association au méthotrexate : traitement de l'uvéite antérieure chronique non infectieuse associée à une arthrite juvénile idiopathique chez l'enfant à partir de 2 ans et l'adolescent, en cas de réponse insuffisante ou d'intolérance au traitement conventionnel ou pour lesquels un traitement conventionnel est inapproprié.
Solution injectable (injection).
Solution claire ou légèrement opalescente, incolore à jaune-brunâtre pâle.
Hulio
40 mg, solution injectable en flacon à usage unique (verre de type I)
muni d'un bouchon en caoutchouc (caoutchouc butyle laminé en
fluoropolymère), avec sertissage en aluminium et d'une capsule amovible.
Chaque boîte contient 1 flacon (0,8 mL de solution stérile), 1 seringue
stérile vide, 1 aiguille stérile, 1 adaptateur pour flacon stérile et 2
tampons d'alcool.
Conditionnement multiple contenant 2 flacons (2 boîtes de 1).
Un flacon unidose de 0,8 mL contient 40 mg d'adalimumab.
L'adalimumab est un anticorps monoclonal humain recombinant produit dans des cellules ovariennes de hamster chinois.
Excipient(s) à effet notoire :
Chaque flacon contient 38,2 mg de sorbitol (E420).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Glutamate monosodique
Sorbitol (E420)
Méthionine
Polysorbate 80
Acide chlorhydrique (pour l'ajustement du pH)
Eau pour préparations injectables